| |
Учебный сайт Юдиной А.С. |
Главная |
Обо мне |
Семестры |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Сборка генома de novo. В данном задании предлагалось поработать с проектом по секвенированию бактерии Buchnera aphidicola. Это бактерия относится к группе протеобактерий, является грамотрицательной и считается родcтвенником современных энтеробактерий. [1]
I часть. Подготовка чтений.Перед началом работы все чтения были обработаны программой trimmomatic. Били удалены адаптеры и плохие буквы с концов, а также чтения с длиной меньше 30 нуклеотидов (так как известно, что в среднем длина 36 нуклеотидов). Все адаптеры для Illumina были собраны в один файл adapters.fasta. Все команды, выполненные в ходе очистки, сведены в таблицу 1. Таблица 1
II часть. Подготовка k-меров.Подготовка k-меров производилась в программе velveth (подпрограмма программы velvet). Эта программа строит хаш-таблицы и создает в отдельной директории два файла - Sequences и Roadmaps, необходимые для работы программы velvetg. Таблица 2
III часть. Сборка на основе k-меров.На данном этапе работы для обработки файлов из предыдущей части используется программа velvetg. Она строит граф де Брайна, граф ориентирован, в его вершинах расположены последовательности символов длины n. Граф отражает пересечения между последовательностями символов. Таблица 3
Граф имеет 1514 вершин и N50 = 67095. Описание контигов с максимальной длиной представлено в таблице 4. Таблица 4
Теперь требовалось определить есть ли контиги с аномально большим или маленьким покрытием, для этого были вычислены наиболее употребляемые средние значения в программе Excel.
Таблица 5
IV часть. Анализ.Теперь требуется сравнить нашу сборку с уже собранной хромосомой того же организма (AC CP009253). Для этого был использован megablast со стандартными параметрами. В таблице 6 представлены результаты работы megablast, основные показатели представлены для самой большой находки Таблица 6
* Контиг отложился частично в начало и частично в конец генома, но это одна находка. Контиг ID 1
 Можно сделать вывод, что относительно положения в геноме данный контиг инвертирован. Контиг ID 7
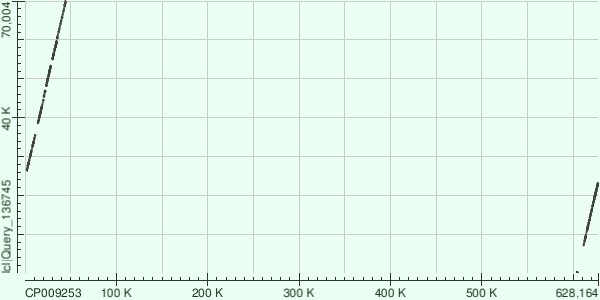 Так как геном бактерии ковалентно замкнут, такрой результат мы могли получить в результате тогго, что при секвенировании референсного генома начало было выбрано в другом месте, нежели при секверинровании нашего контига. Контиг ID 9
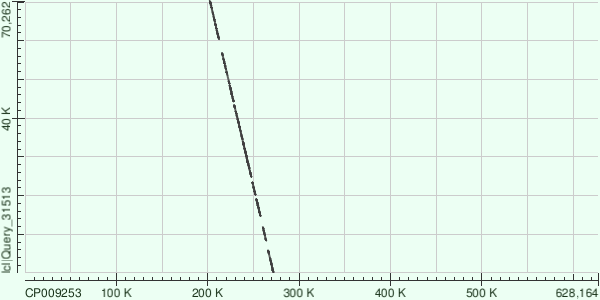 Как и в случае с первым рассматриваемым контигом, последовательеность инвертирована относительно генома. Общий вывод по всем контигам: находки выравниваний контига и генома расподожены на геноме подряд и имеют хороший вес и E-value, указывающее на достоверность этих находок. Источники[1] wikipedia.org |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
© Юдина Анастасия, 2016