|
Home page Term 1 Term 2 Term 3 About me Faculty website |
De novo genome assemblyReads preparationFirst, the .fastq.gz was unzipped with gunzip, which yielded a 1808 Mb file, and the adapters were concatenated:cat /P/y16/term3/block3/adapters/*.fa adapters.fastaThen, the adapters were deleted from the reads set using trimmomatic: java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 reads14.fastq reads14_na.fastq ILLUMINACLIP:adapters.fasta:2:7:7The result was this (and a 1807 Mb file): Input Reads: 17756177 Surviving: 17750402 (99,97%) Dropped: 5775 (0,03%)Afterwards, trimmomatic was employed again to delete bad quality (lower than 20) ends of the reads and delete overly short ones (less than 30 nt): java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 reads14_na.fastq reads14_na_trimmed.fastq TRAILING:20 MINLEN:30This is the result (and a 1173 Mb file): Input Reads: 17750402 Surviving: 11913544 (67,12%) Dropped: 5836858 (32,88%) VelvethVelveth is a program for preparation of kmers needed to assemble the genome (we are dealing with Buchnera aphidicola's genome). This is how it was usedvelveth kmers_velveth 29 -short -fastq reads14_na_trimmed.fastqIt returns a directory (kmers_velveth in this case) containing the output files. VelvetgVelvetg is a program for assembly of the genome based on velveth's output (kmers). This is how it was launched:velvetg kmers_velvethN50 of the assembly was 13439 (as presented in velvetg's Log file). The three longest contigs:
* The coverage presented is from the short1_cov column Sequence of contig 1 Sequence of contig 13 Sequence of contig 8 The median coverage was 39.0269 , and there were 5 contigs with anomalous coverages: three had extremely low ones (2.7, 2.9, and 4.0) and two had high ones (269.2 and 274.6). It is worth noticing that all five were among the shortest contigs returned by the program. All results here were obtained via MS Excel. Megablast and analysis
* This is actually the count of gap openings. The information was obtained from the corresponding hit tables using MS Excel. Here are the dot matrix views for all the alignments: Contig 1: 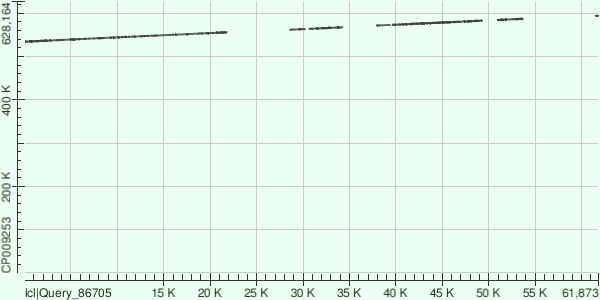 Contig 13: 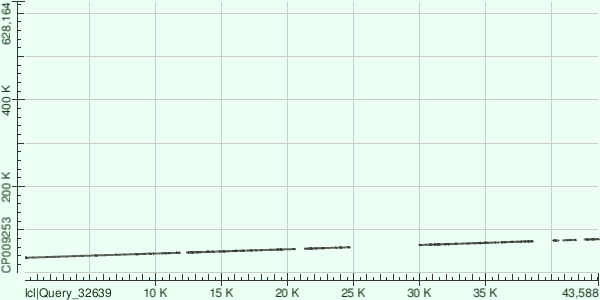 Contig 8: 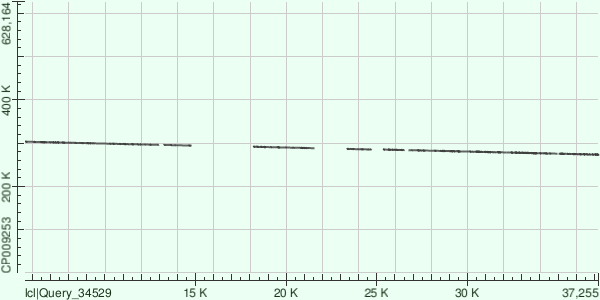 Overall, the genome that the contigs were aligned to (GenBank/EMBL AC — CP009253) contained plenty of indels compared to the strain of Buchnera aphidicola that had been sequenced. |
© Stanislav Tikhonov, 2018