|
|
В качестве референсной последовательности, на которую картировались
исследуемые чтения, использовалась хромосома 8 H. sapiens.
Все причастные файлы находятся в рабочей директории /nfs/srv/databases/ngs/
stepan_puhov/pr11 в соответствующих поддиректориях. Для точности описания
команд указывается, в каких директориях производился их запуск.
|
Подготовка референса
Для картирования ридов референсная последовательность индексируется.
Индекс последовательности - сжатый формат последовательности, по которому
можно быстро проводить поиск подпоследовательностей.
Для индексирования референса была использована программа
hisat2-build:
Команда: (в директории reference) hisat2-build chr8.fasta chr8
Вывод программы: 8 файлов вида сhr8.<file number>.ht2,
вместе составляющие индекс референсной последовательности
|
Анализ качества чтений
Для оценки качества чтений была использована программа
FastQC:
Команда: (в директории reads) fastqc --extract chr8.fastq
Вывод программы: страница с
отчётом
Из первого раздела отчёта FastQC видно, что имеется
всего 8367 чтений,
из которых ни одно не помечено как "чтение плохого качества" и длина которых
составляет от 34 nt до 100 nt, кодировка качества определена как Phred+33
(Sanger/Illumina 1.9).
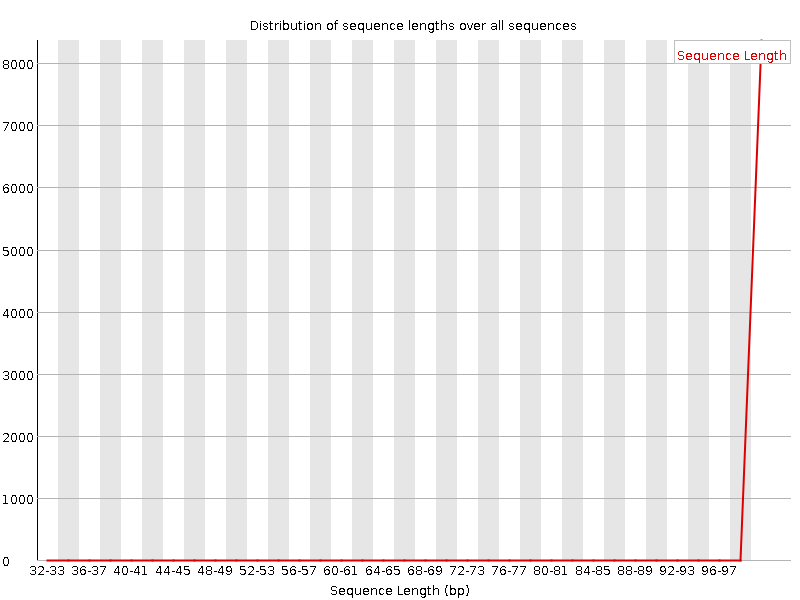
Несмотря на большой размах длин, из раздела "Sequence length distribution"
отчёта видно, что практически все чтения всё же имеют длину 100 nt, однако
раздел выдаёт предупреждение (выдаётся, если не все чтения одной длины) из-за
наличия чтений меньшей длины (видимо, в очень малом количестве):
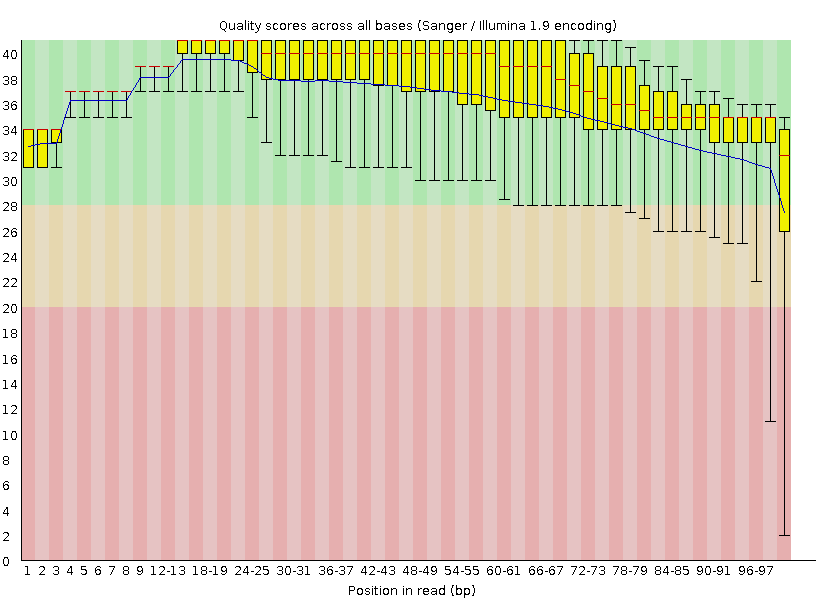
В разделе "Per base sequence quality" отчёта представлена диаграмма типа
boxplot распределения качества определения нуклеотида для каждой позиции
в чтении: поскольку максимальная длина чтения − 100 nt, показано 100
позиций, для каждой позиции жёлтый прямоугольник представляет интерквартильный
интервал качества, усы − квантиль уровня 0.1 и квантиль уровня 0.9,
красная полоса − медиану качества, синяя линия − ожидаемое
качество.
Видно, что большинство имеют хорошее или приемлимое качество на всём своём
протяжении, хотя ближе к 3'-концу оно несколько ухудшается: вплоть до 78 nt
в 90% случаев определение выбранного нуклеотида будет иметь хорошее качество
(Q > 28) и, только начиная примерно с 97 nt, доля случаев, в которых выбранный
нуклеотид определяется с плохим качеством (Q < 20), начинает превышать 10%:
|
Очистка чтений и повторный контроль их качества
Для очистки чтений с их 3'-конца были удалены все нуклеотиды, качество которых
ниже 20, и после оставлены только чтения с длиной не менее 50 nt, была
использована программа Trimmomatic:
Команда: (в директории reads)
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/
trimmomatic-0.30.jar
SE -phred33 chr8.fastq chr8_trimmed.fastq TRAILING:20 MINLEN:50
Вывод программы: файл с очищенными чтениями chr8_trimmed.fastq и
отчёт о работе, выданный в stder
Далее был проведён повторный контроль качества чтений программой
FastQC:
Команда: (в директории reads)
fastqc --extract chr8_trimmed.fastq
Вывод программы:
страница с отчётом
Из отчёта Trimmomatic видно, что после триммирования осталось
всего 8227 чтений, при этом было удалено 140 чтений (1.67%).
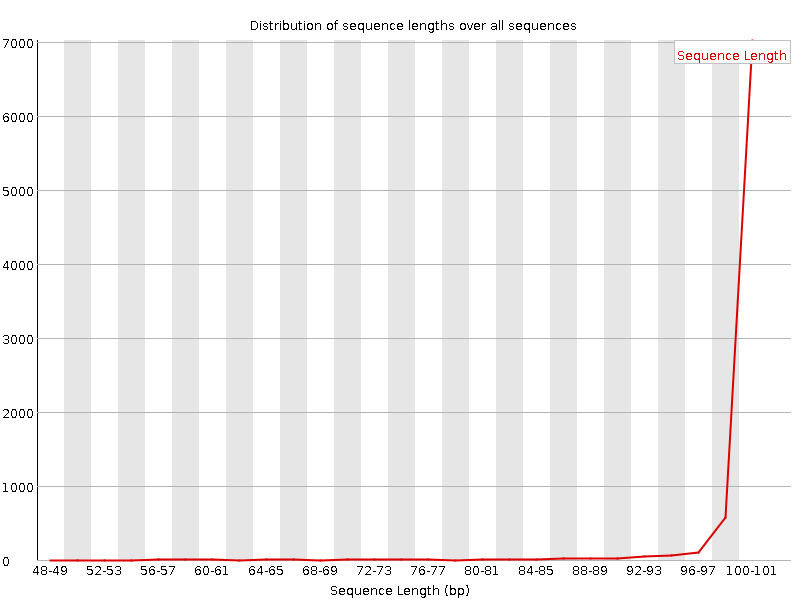
Из анализа FastQC видно, что теперь все чтения имеют длину от 50 nt
до 100 nt, то есть, видимо, изначально существовали короткие чтения разной
длины, в том числе и более 50 nt, в разделе "Sequence length distribution"
теперь заметны чтения длиной от 90 nt:
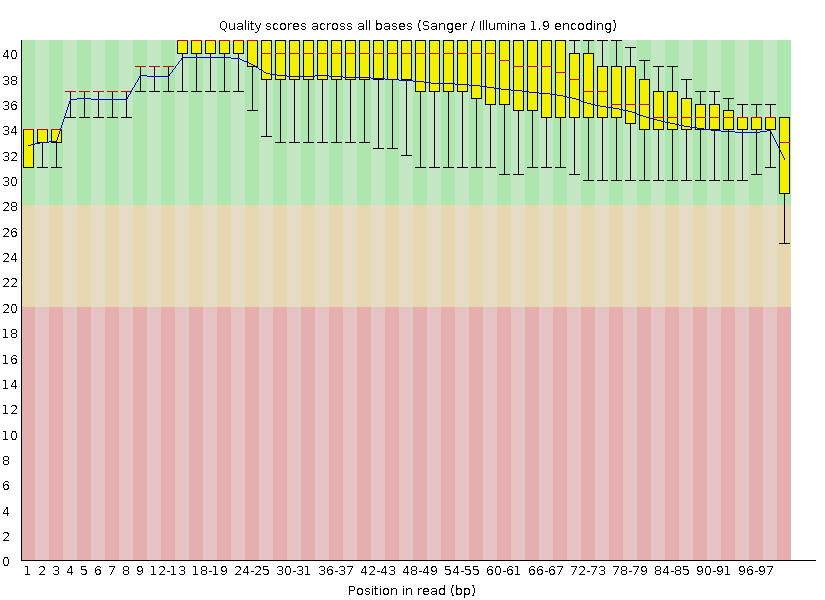
В разделе "Per base sequence quality" можно видеть, что качество определения
нуклеотидов улучшилось для всех позиций чтений, произошло это из-за удаления
или триммирования коротких чтений, которые могли несколько
ухудшать качество в начальных и средних позициях; теперь качество определения
нуклеотидов хорошее практически во всех случаях для позиций до 97 nt, в
остальных позициях качество хорошее чуть больше, чем в 75% случаев:
Из-за изначально хорошего качества чтений триммирование в данной ситуации не
являлось необходимым, но, учитывая заметное улучшение качества чтений, было
желательным.
|
Картирование чтений и анализ выравнивания
Картирование триммированных чтений на индексированную референсную
последовательность было осуществлено программой hisat2:
Команда: (в директории alignments)
hisat2 --no-softclip --no-spliced-alignment --summary-file mapping_report.txt
-x ../reference/chr8 -U ../reads/chr8_trimmed.fastq -S chr8.sam
Вывод программы: SAM-файл с картированными чтениями chr8.sam
и отчёт картирования
Из отчёта картирования программы
hisat2 видно, что из 8227 данных программе чтений 30 не картировались,
а все остальные 8197 чтений картировались ровно 1 раз, таким образом было
картировано 99.64% чтений.
Программа samtools flagstat (команда samtools flagstat chr8.sam)
выдала отчёт, где все 8227 чтений и, соответственно, 8197 картированных чтений
отмечены как прошедшие проверку качества. При быстром просмотре получившегося
SAM-файла видно, что практически все чтения имеют mapping quality = 60 −
очень высокое качество (вероятность ошибки в координате картирования равна
10-6). Можно заключить, что картирование имеет высокое качество.
Далее SAM-файл был переведён в индексированный отсортированный BAM-файл с
помощью программ Samtools (все команды в директории alignments):
-
Перевод в BAM-формат: samtools view -b -o chr8.bam chr8.sam
-
Сортировка BAM-файла по координатам картирования: samtools sort chr8.bam
chr8_sorted, выдача: chr8_sorted.bam
-
Индексирование отсортированного BAM-файла: samtools index
chr8_sorted.bam, выдача: chr8_sorted.bam.bai
|
Поиск SNP и инделей
Для поиска отличий картированных чтений от референса был создан файл
формата bcf с помощью samtools mpileup:
Команда: (в директории polymorph)
samtools mpileup -u -f ../reference/chr8.fasta -o chr8.bcf
../alignments/chr8_sorted.bam
Вывод программы: бинарный файл chr8.bcf с отличиями
картированных чтений от референса
Файл формата bcf был переведён в файл формата vcf с помощью
bcftools call:
Команда: (в директории polymorph)
bcftools call -cv -o chr8.vcf chr8.bcf
Вывод программы: текстовый файл chr8.vcf с отличиями
картированных чтений от референса
Из полученного vcf файла видно, что исследуемый
образец несёт всего 100
полиморфизмов, из которых 5 инделей и 95 snp.
Описание 3 из этих полиморфизмов:
| Полиморфизм 1 | Полиморфизм 2 | Полиморфизм 3 |
| Координата | 27454785 | 27464081 | 116424270 |
| Тип полиморфизма | делеция | snp | инсерция |
| Последовательность в референсе | TAATGAA | C | A |
| Последовательность в чтениях | TAA | T | AC |
| Глубина покрытия в этом месте | 5 | 17 | 28 |
| Качество чтений в этом месте | 58.4663 | 111.008 | 57.456 |
Стастистика качества чтений и покрытий участков, содержащих найденные
полиморфизмы:
| Среднее значение | Минимальное значение | Первый квартиль | Медиана | Второй квартиль | Максимальное значение |
| Качество чтений | 66.05637 | 3.01618 | 7.79993 | 11.34290 | 113.37025 | 225.00900 |
| Глубина покрытия | 14.87 | 1 | 1 | 2 | 16.25 | 134 |
|
Аннотация snp
Для аннотации snp файл формата bcf, содержащий отличия картированных чтений
от референса, был переведён в формат vcf без учёта инделей с помощью
bcftools call:
Команда: (в директории polymorph)
bcftools call -cv -V indels -o chr8_snp.vcf chr8.bcf
Вывод программы: файл chr8_snp.vcf, содержащий только
однонуклеотидные замены
Далее полученный vcf файл с snp был переведён в формат, подходящий для
программы аннотации ANNOVAR, с помощью convert2annovar.pl:
Команда: (в директории polymorph)
convert2annovar.pl -format vcf4 chr8_snp.vcf -outfile snp.avinput
Вывод программы: файл snp.avinput, пригодный для работы
программы аннотации
Однонуклеотидные полиморфизмы из файла snp.avinput были аннотированы
по 5 базам данных с помощью annotate_variation.pl:
-
Аннотация, основанная на генной разметке, по базе refgene:
annotate_variation.pl -dbtype refGene -buildver hg19 -out
refgene/snp_refgene snp.avinput /nfs/srv/databases/annovar/humandb.old
-
Аннотация, основанная на фильтрации, по базе dbsnp:
annotate_variation.pl -filter -dbtype snp138 -buildver hg19 -out
dbsnp/snp_dbsnp snp.avinput /nfs/srv/databases/annovar/humandb.old
-
Аннотация, основанная на фильтрации, по базе 1000 genomes:
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out
1000genomes/snp_1000genomes snp.avinput
/nfs/srv/databases/annovar/humandb.old
-
Аннотация, основаная на разметке регионов генома, по базе GWAS:
annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19
-out gwas/snp_gwas snp.avinput /nfs/srv/databases/annovar/humandb.old
-
Аннотация, основанная на фильтрации, по базе Clinvar:
annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19
-out clinvar/snp_clinvar snp.avinput /nfs/srv/databases/annovar/humandb.old
Из аннотации по базе refgene можно сделать следующие выводы:
-
База данных refgene вообще делит snp на 9 категорий:
intergenic, intronic, exonic, UTR5, UTR3,
ncRNA, splicing, upstream и downstream.
snp исследуемого образца попали в только 4 категорий:
| Категория | Количество snp |
| intergenic | 17 |
| intronic | 60 |
| exonic | 5 |
| UTR3 | 13 |
-
snp исследуемого образца попали в следующие 3 гена:
| Название гена | Количество snp |
| CLU | 10 |
| HNF4G | 22 |
| TRPS1 | 46 |
-
snp исследуемого образца привели к следующим заменам в экзонах и белках
(всего 5 замен в экзонах и 2 замены в белках):
| Координата на хромосоме | Ген | Замена в экзоне | Замена в белке |
| 27462481 | CLU
(ген на комплементарной цепи по отношению к последовательности референса) | T → C | синонимичная
(сохраняется His) |
| 76452313 | HNF4G | G → A | Ser → Gln |
| 76468228 | HNF4G | G → A | синонимичная
(сохраняется Leu) |
| 76468282 | HNF4G | G → A | Met → Ile |
| 116631902 | TRPS1
(ген на комплементарной цепи по отношению к последовательности референса) | G → T | синонимичная
(имеется 3 варианта сплайсинга, во всех случаях сохраняется Pro) |
Из аннотации по базе dbsnp видно, что 77 snp исследуемого образца
имеют rs.
Из аннотации по базе 1000 genomes можно сделать выводы о частоте
найденных полиморфизмов. Некоторые полиморфизмы имели заявленную частоту
равную 1, что кажется бессмысленным, для остальных были подсчитаны
характеристики распределения:
| Среднее значение | Минимальное значение | Нижний квартиль | Медиана | Верхний квартиль | Максимальное значение |
| 0.40722 | 0.00499 | 0.05481 | 0.51337 | 0.61541 | 0.79712 |
В целом, можно видеть большинство snp найденных достаточно распостранены в
популяции; отдельно было подсчитано, что редких snp (частота < 0.005) было
найдено всего 3.
Аннотация по базе GWAS показывает, что из 95 snp исследуемого образца
4 snp несут клиническое значение:
2 snp в интронах гена CLU ассоциированы с повышенным риском болезни Альцгеймера,
1 snp в 3'-UTR гена HNG4G влияет на уровнь мочевой кислоты и
1 snp в интроне гена TRPS1 ассоциирован с уровнем холестерола, связанного
с липопротеинами высокой плотности.
Аннотация фильтрацией по базе Clinvar не даёт никаких
результатов − все 95 snp не прошли критерии фильтрации.
|
|
|