|
|
Произведена сборка контигов на основании одноконцевых прочтений, полученных
секвенированием транскриптома растения Arabidopsis thaliana (файл D).
|
Методы
Все полученные файлы хранятся в рабочей директории
/nfs/srv/databases/ngs/stepan_puhov/pr14
в соответствующих поддиректориях.
Далее представлена таблица, отражающая последовательность действий и
использованных команд (для точности описания указывается, в каких директориях
производился запуск соответствующих команд):
| Действие | Команда |
| Разархивирование чтений | (в директории reads)
gunzip D.fastq.gz |
| Создание файла с адаптерами | (в директории reads)
cat /P/y18/term3/block3/adapters/* > adapters.fasta |
| Очистка чтений от адаптеров | (в директориии reads)
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/ trimmomatic-0.30.jar SE -phred33 D.fastq reads.fastq ILLUMINACLIP:adapters.fasta:2:7:7 |
| Триммирование чтений | (в директории reads)
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/ trimmomatic-0.30.jar SE -phred33 reads.fastq reads_trimmed.fastq SLIDINGWINDOW:5:28 MINLEN:32 |
| Оценка качества чтений до триммирования | (в директории reads)
fastqc --extract reads.fastq |
| Оценка качества чтений после триммирования | (в директории reads)
fastqc --extract reads_trimmed.fastq |
| Создание 31-меров из чтений | (в директории pr14)
velveth assemblage 31 -short -fastq reads/reads_trimmed.fastq |
| Построение графа де Брёйна и сборка контигов | (в директории pr14)
velvetg assemblage |
| Определение длины 3 самых длинных контигов | (в директории assemblage)
grep -E '^>' contigs.fa | sed -r 's/.+length_([0-9]+)_.+/\1/' | sort -g | tail -n 3 |
| Поиск 3 самых длинных контигов | (в директории assemblage)
grep -E 'length_([length1]|[length2]|[length3])_' contigs.fa,
где [length1-3] − длины 3 самых длинных контигов |
| Определение наибольшего и наименьшего покрытия | (в директории assemblage)
grep -E '^>' contigs.fa | sed -r 's/.+_cov_([0-9]+\.[0-9]+)$/\1/' | sort -g | sed -r -n -e '1p' -e '$p' |
| Поиск контига с наибольшим покрытием | (в директории assemblage)
grep -E '_cov_[maxcov]$' contigs.fa,
где [maxcov] − максимальное покрытие |
| Поиск контига с наименьшим покрытием | (в директории assemblage)
grep -E '_cov_[mincov]$' contigs.fa | head -n 200,
где [mincov] − минимальное покрытие |
| Подсчёт контигов с минимальным покрытием | (в директории assemblage)
grep -E -c '_cov_[mincov]$' contigs.fa |
| Получение последовательностей самого длинного контига и контигов с наибольшим и наименьшим покрытием | (в директории assemblage)
seqret @3contigs.list 3contigs.fa,
где файл 3contigs.list содержит USA адреса в файле contigs.fa для выбранных 3 контигов |
| Аннотация контигов | (на сайте NCBI)
Поиск blastn со стандартными параметрами по базе Nucleotide collection |
|
Подготовка чтений
Очистка чтений от адаптеров и триммирование были произведены с помощью
программы Trimmomatic:
-
Возможные последовательности адарптеров были собраны в файл
adapters.fasta, команда ILLUMINACLIP:adapters.fasta:2:7:7
использована для очистки чтений.
Из отчёта программмы видно, что
1333 последовательности (0.03%) из файла с чтениями были распознаны как
остатки адаптеров и удалены.
-
Триммирование очищенных от адаптеров чтений было осуществленно методом
скользящего окна длиной 5 nt с минимальным допустимым качеством 28 −
отрезается 3'-конец последовательности перед первым квинтетом нуклеотидов,
чьё среднее качество ниже Q = 28 (команда SLIDINGWINDOW:5:28); были
сохранены только те тения, длина которых после удаления 3'-конца была не
меньше 32 nt (команда MINLEN:32).
Из отчёта программы видно, что
до триммирования было 3868536 чтений (в файле размером 990.93 Mb), после
триммирования сохранилось 91.31% чтений, то есть 3532428 чтений (в файле
размером 848.91 Mb). Визуализация программой FastQC показывает, что
качество чтений значительно улучшилось после триммирования:
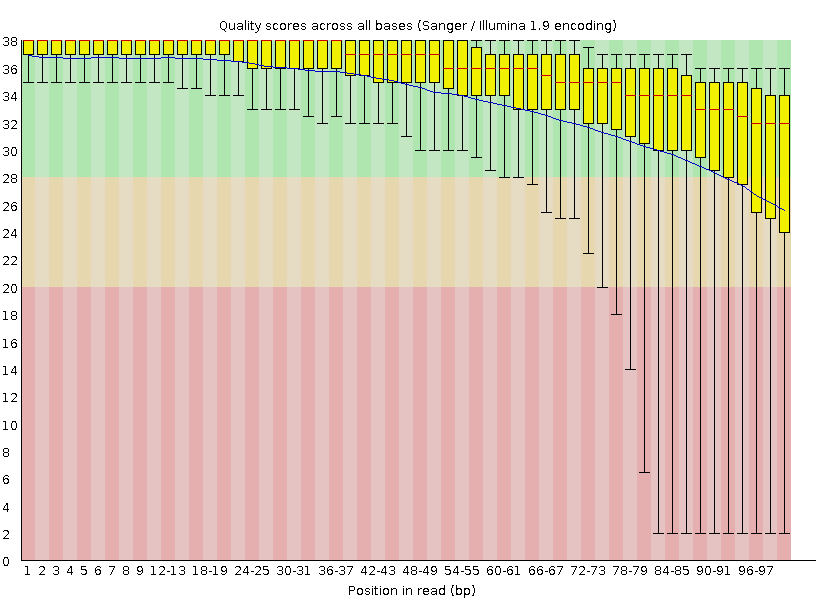
Качество чтений без адаптеров до триммирования
|
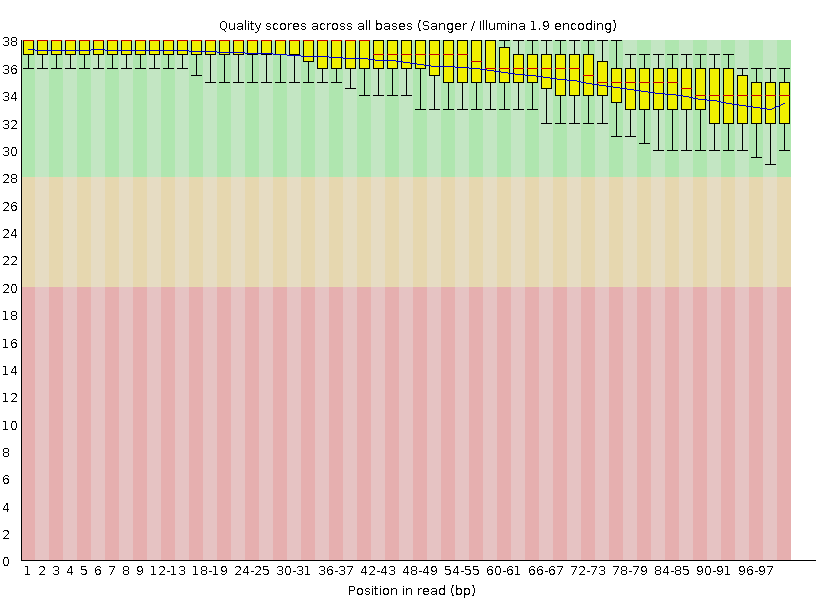
Качество чтений без адаптеров после триммирования
|
|
Сборка контигов
Сборка контигов на основании триммированных чтений была произведена
программой Velvet:
-
Команда velveth была использована для создания 31-меров на основании
чтений.
-
Построение графа де Брёйна на полученных 31-мерах, его упрощение и отбор
контигов (выбирались узлы окончательной версии графа с длиной не менее 2
31-меров) осуществлены командой velvetg.
Из краткого отчёта работы программы можно увидеть,
что для отобранных контигов N50 (минимальная длина контига из набора самых
длинных контигов, вместе составляющих 50% общей длины всех контигов) равняется
67 nt.
Поскольку в отчёте stats.txt есть информация о всех узлах окончательной версии
графа (в том числе слишком коротких, не попавших в контиги), информация о длинах
и покрытиях контигов (во избежании неприятной автору работы с Excel) была
получена из файла contigs.fa с помощью программ grep, sed и
sort
(используется
специальное представление
длины и покрытия в k-мерах):
|
Аннотация контигов
Для аннотации самого длинного контига и контигов с наименьшим и наибольшим
покрытиями был произведён поиск последовательностей этих контигов в базе
данных Nucleotide collection алгоритмом blastn с параметрами по умолчанию.
Поиск по всем организмам, а не только по записям для исследуемого организма,
и использование стандартного blastn оправданы тем, что это может позволить
обнаружить контиги, собранные неверно за счёт различных контаминаций.
Отчёт BLAST поиска самого длинного
контига.
Около половины находок представлено 4 различным участкам генома
A. thaliana, остальные находки принадлежат другим растениям семейства
Brassicaceae; ни одна находка не имеет 100% покрытия запроса.
Из графической выдачи BLAST (представлены
только находки, принадлежащие A. thaliana) видно, что
контиг состоит из 2 независимых участков: 5'-концевого участка (примерно
220 nt первых нуклеотидов), относящегося к гену 5 хромосомы белка
из семейства P-loop ATPases, и 3'-концевого участка (все остальные нуклеотиды
контига), относящегося к 3 паралогичным генам, DNA-binding storekeeper-like
protein хромосомы 5 и DNA-binding storekeeper protein-related transcriptional
regulator хромосом 2 и 4.
Из анализа полученных выравниваний можно сделать
вывод, что контиг ошибочно собран из 3 участков: участок 1-221 nt принадлежит
мРНК гена P-loop ATPase, образующей нуклеотидсвязывающий домен неустановленного
ABC-транспортёра, с хромосомы 5 (выравнивание,
направление цепей совпадает),
участок 218-518 nt принадлежит мРНК гена DNA-binding storekeeper
protein-related transcriptional regulator с хромосомы 2
(выравнивание, контиг имеет обратную
цепь), участок 463-665 nt
принадлежит мРНК гена DNA-binding storekeeper-like protein с хромосомы 5
(выравнивание, контиг имеет обратную
цепь). Участки определялись, как части выравниваний со 100% совпадаением и
без гэпов, они имеют перекрывания из-за того, что соответствующие гены имеют
совпадающие участки; упомянутый паралог из хромосомы 4 имеет несовпадения с
контигом по всей длине выранивания, но по сути 31-меры из прочтений мРНК могли
быть включены в контиг.
Отчёт BLAST поиска контига с
наибольшим покрытием.
Лучшие находки здесь составляют группу одинаковых находок, имеющих по одному
выравниванию с контигом без гэпов, со 100% сходством и полным покрытием.
Таким образом, данный контиг несомненно является участком мРНК гена
неописанного трансмембранного белка с хромосомы 5 A. thaliana
(выравнивание).
Отчёт BLAST поиска контига с
наименьшим покрытием.
Большая часть находок принадлежит растениям из группы Pentapetalae,
из них значительная часть находок представлена 3 различными участками
генома A. thaliana, остальные находки принадлежат различным животным,
споровику и даже кренархее, но имееют слишком высокое E-value (от 1.5).
Из графической выдачи BLAST
(отображены только находки, принадлежащие A. thaliana) можно сделать
вывод, что контиг ошибочно собран из 2 участков: участок 1-30 nt принадлежит
мРНК гена DEAD-box хеликазы с хромосомы 1
(выравнивание),
участок 21-75 nt принадлежит мРНК гена компонента протеасомы с хромосомы 3
(выравнивание).
Второй участок представлен наиболее правдоподобной находкой, имеющей 1
выравнивание с E-value 1e-13, 2 гэпами, 96% идентичности и 73% покрытием
запроса.
|
|
|
{kind=link}
{kind=link}