|
Предсказание вторичной структуры РНК
Файл с последовательностью tRNAPro(CGG) бактерии
Thermus thermophilus (PDB ID 1H4S) в fasta-формате 1H4S.fasta был
загружен со страницы PDB.
Замечание: В pdb-файле дана неполная последовательность тРНК -
с 4 по 69 нуклкотид из 77 нуклеотидов в fasta-файле. Хуже того, имеется
несколько нетрадиционный способ нумерации в pdb-файле - после 17 нуклеотида
идёт 17^A нуклеотид, а только за ним 18, поэтому нумерация после 17 нуклеотида
отличается в выводе einverted, в отчёте использована нумерация из файла
pdb. Тимидин и псевдоуридин в fasta-файле обозначены как уридин.
С помощью программ einverted, ищущей комплементарные участки цепи, и
RNAfold, выполняющей алгоритм Зукера предсказания вторичной структуры
РНК, была предсказана вторичная структура данной тРНК, результаты предсказаний
сравнены с выводами программы find_pair на основании структуры PDB:
-
Программа einverted предсказывает вторичную структуру РНК, ища
комплементарные участки цепи, учитывая при этом только канонические пары
нуклеотидов. Спирали отбираются по счёту (параметр -threshold),
сформированного баллами за нахождения в спирали комплементарных пар
(параметр -match), выпетливаний (параметр -gap) или внутренних петель
(параметр -mismatch).
Данный метод предсказания оказался довольно неточным: при различных сочетаниях
параметров предсказывалась одна из 4 структур (или не предсказывалось
ничего):
-
только Acceptor-stem (например, при запуске с параметрами
-gap 10 -threshold 20 -match 7 -mismatch -10),
-
участок TψC-stem с большим
количеством лишних пар (например, при запуске с параметрами
-gap 10 -threshold 20 -match 8 -mismatch -10),
-
вся молекула со вторичной
структурой типа helix-bulged helix-helix-loop c правильно определёнными
Acceptor-stem и Anticodon-stem (например, при запуске с параметрами
-gap 10 -threshold 20 -match 9 -mismatch -10),
-
похожа на структуру 3, в Anticodon-loop лишние пары (например, при запуске с
параметрами -gap 10 -threshold 20 -match 10 -mismatch -10).
Лучшей из этих структур можно считать третью:
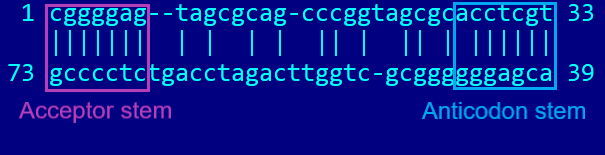
-
Программа RNAfold для предсказание вторичной структуры РНК реализует
алгоритм поиска структуры с минимальной свободной энергией образования (MFE
structure). Параметр --MEA позволяет помимо MFE structure рассчитывать
вероятность образования пары для каждого основания и отображать (в скобочной
записи) структуру, в которой отмечены пары с достаточно большой (чем больше
значение параметра, тем меньше порог) вероятностью образования пары.
Метод предсказания с первого запуска (с параметром --MEA=1 по умолчанию) дал
довольно близкий к действительности результат: все стебли предсказаны верно,
кроме предсказания лишней пары внутри Anticodon-loop и отсутствия
пары AG − первой пары Anticodon-stem, несмотря на
параметр --nsp -AG (учитывает возможность неканонической пары AG).
Увеличения параметра --MEA (что должно позволять предсказывать пару между
нуклеотидами, имеющими невысокую вероятность образования таковой) не добавляет
первую пару в антикодоновый стебель, но предсказывает лишние 2 пары
в D-loop.
Таким образом, лучшая структура:
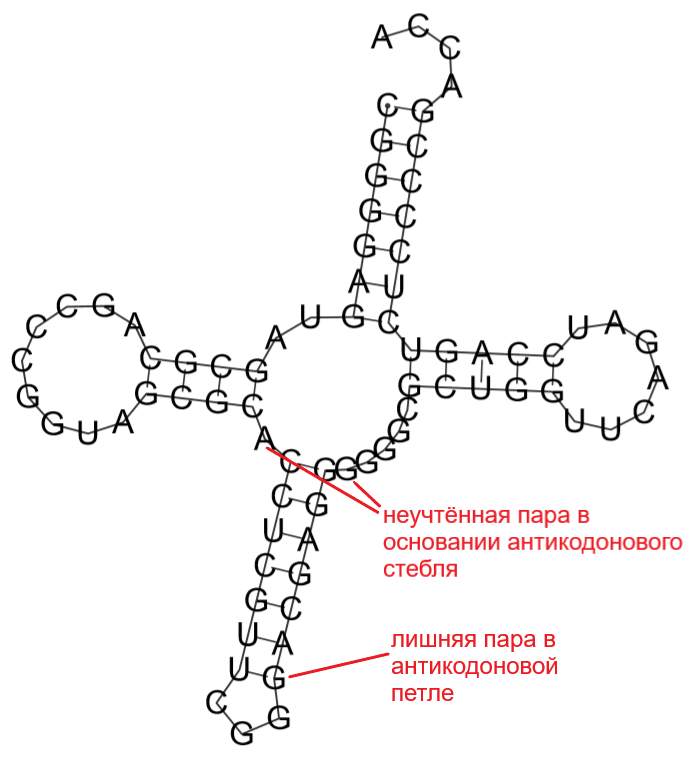
Сравнение со структурой pdb:
| Участок структуры | Позиции в структуре (по результатам find_pair из pdb) | Результаты предсказания с помощью einverted | Результаты предсказания с помощью RNAfold |
| Acceptor-stem | 5'-4-7-3'
5'-66-69-3'
Всего 4 пары, из них 0 неканонических | предсказано 4 пары из 4 в pdb + 3 пары, отсутствующие в pdb | предсказано 4 пары из 4 в pdb + 3 пары, отсутствующие в pdb |
| D-stem | 5'-10-13-3'
5'-22-25-3'
Всего 4 пары, из них 0 неканонических | — | предсказано 4 пары из 4 реальных |
| Anticodon-stem | 5'-26-32-3'
5'-38-44-3'
Всего 7 пар, из них 1 неканоническая | предсказано 6 пар из 7 реальных | предсказано 6 пар из 7 реальных + 1 лишняя пара |
| TψC-stem | 5'-49-53-3'
5'-61-65-3'
Всего 5 пар, из них 1 неканоническая | — | предсказано 5 пар из 5 реальных |
| Общее число канонических пар нуклеотидов | 18 | 13 | 21 |
(полученные файлы хранятся в директории
~stepan_puhov/term3/block1/pr3/rnastructure)
|
Поиск ДНК-белковых контактов
ДНК-белковые контакты в комплексе домена MH1 белка SMAD3 человека с ДНК
(PDB ID 1MHD) были сначала визуализированы с помощью JMol, а затем
с помощью программы nucplot.
Файл 1MHD.pdb был загружен с сайта PDB, затем переведён программой
remediator в старый формат (PDBv2.3) 1MHD_old.pdb для
дальнейшей обработки программмой nucplot, все дальнейшие операции
проводились с этим файлом (т.е. скрипты JMol были
написаны под старый формат).
Файл содержит 2 одинаковых белка, образующих комплексы с разными (отдалёнными)
участками ДНК, исследовалось взаимодействие только одного белка с ДНК −
цепь A в pdb.
(полученные файлы хранятся в директории
~stepan_puhov/term3/block1/pr3/complexstructure)
|
Визуализация молекулы ДНК в JMol
Написаны 2 скрипта для JMol:
-
Скрипт,
задающий множество атомов кислорода дезоксирибозы как "set1",
множество атомов килорода фосфатных остатков ДНК − как "set2" и
множество атомов азота в азотистых основаниях − как "set3".
-
Скрипт,
последовательно показывающий всю исследуемую структуру (цепь A и ДНК); только
молекулу ДНК в проволочной модели; в этой модели выделены шариками только
атомы из set1; выделены шариками только атомы из set2; выделены шариками
только атомы из set3.
|
|
Визуализация контактов в JMol
Определим атомы кислорода и азота как полярные, а атомы углерода, фосфора и
серы − как неполярные.
Определим полярный контакт как пару полярных атомов ДНК и белка,
находящихся на расстоянии меньшим 3.5Å, и неполярный контакт
− как пару неполярных атомов ДНК и белка, находящихся на расстоянии
меньшим 4.5Å (при таком определении один атом может участвовать сразу
в нескольких контактах).
Был написан скрипт
для JMol последовательно визуализирующий для исследуемой структуры
атомы дезоксирибозы; атомы фосфатных остатков ДНК; атомы азотистых оснований,
направленные в большую бороздку; атомы азотистых оснований, направленные в
малую бороздку; полярные и неполярные контакты белка с перечисленными
множествами атомов.
Результаты подсчёта этих контактов приведены в таблице:
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 1 | 6 | 7 |
| остатками фосфорной кислоты | 5 | 1 | 6 |
| остатками азотистых оснований со стороны большой бороздки | 6 | 10 | 16 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 0 | 0 |
При данном определении контактов именно полярные контакты будут скорее всего
вносить значимый вклад во взаимодействие белка с ДНК. Видно, что больше всего
полярных контактов белок образует с фосфатными остатками и атомами азотистых
оснований, ориетированными в большую бороздку − с помощью этих контактов
белок связывается с ДНК. Больше всего контактов белок образует с большой
бороздкой, тогда как с малой бороздкой контакты отсутствуют (на визуализации
явно видно, что белок далеко отстоит от малой бороздки) − узнавание
последовательности ДНК происходит по большой бороздке.
|
|
Поиск контактов через nucplot
Визуализация контактов
программой nucplot. Здесь отмечены контакты также для другой
молекулы белка, цепи B; поскольку цепь B в исследуемую структуру не входит,
ниже описываются контакты только для цепи A.
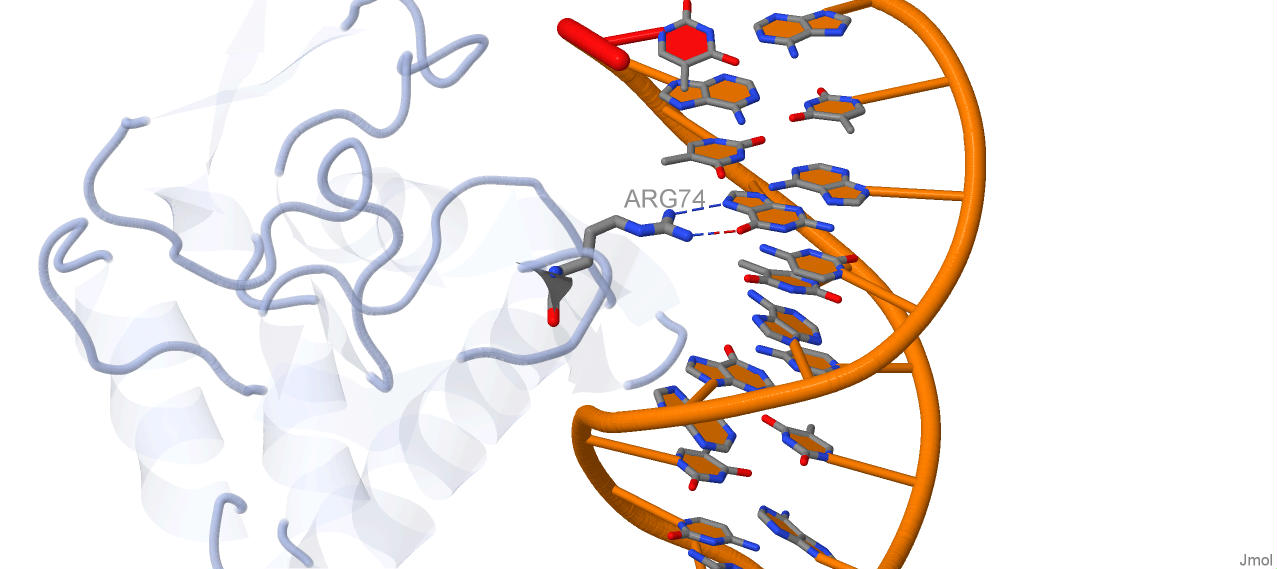
Больше всего контактов c ДНК образует остаток Arg74 − 2 водородные
связи c гуанином (nucplot cчитает, что остаток Lys81 также образует 2
контакта без связей, но JMol рассчитывает 1 из этих контактов как
водородную связь, второй контакт является просто близким положением):
За узнавание ДНК скорее всего отвечают аминокислотные остатки Arg74, образующий
2 водородные связи с гуанином, и Lys81, образующий 1 водородную свзязь с атомом
N7 аденина, а также, возможно, остаток Gln76, ориентирующий Lys81 водородной
связью и, что кажется вероятным из структуры, но не определяется командой
calculate hbonds JMol, может образовывать водородную связь с тем же
аденином по атому N6 (это водородная связь была обозначена чёрным отрезком):
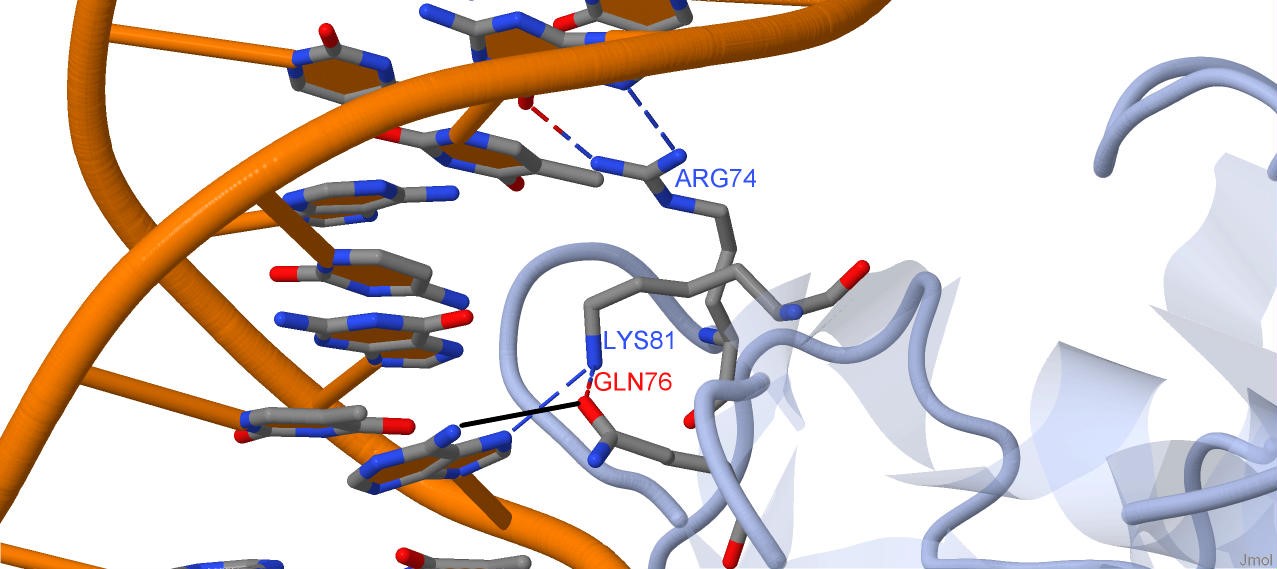
|
|
|
|