|
Аннотация секвенированной последовательности
Для определения принадлежности
консенсусной последовательности ДНК,
полученной анализом хроматограмм сэнгеровского секвенирования в практикуме 6,
был использован поиск алгоритмом blastn с параметрами по умолчанию
по базе Nucleotide collection.
Выбранный алгоритм лучше всего подходит для поиска
гомологов исследуемой нуклеотидной последовательности, поскольку для неё
неизвестно, является ли она белок-кодирующей, а также не гарантируется
присутствие близких гомологов в базе данных. Выбранная база данных позволяет
провести поиск по всем аннотированным последовательностям GenBank, EMBL, DDBJ
и PDB (наиболее широкий вариант поиска).
Из выдачи BLAST можно сделать вывод, что
исследуемая последовательность (запрос) является участком гена, кодирующего
суб'единицу 1 цитохром c оксидазы, так как все находки из 100 находок выдачи
являются участками данного гена, причём уровень покрытия запроса не опускается
ниже 97%, а E-value не превышает 9e-96.
Вследствие высокого покрытия и хорошего E-value таксономию запроса можно
определить, исходя из процента совпадений (%Identity) с находками и общей
вариабельности последовательности данного гена на уровне семейства, рода или
вида. Все 100 находок, кроме 1, принадлежат голожаберникам инфраотряда Doridina
(1 находка принадлежит неопределённому виду группы Gastropoda), при
ранжировании по %Identity все находки с уровнем совпадения выше 97% (они имеют
100% покрытие запроса) принадлежат виду Onchidoris muricata, далее
наблюдается резкий спад уровня совпадения до 87% − можно предположить,
что исследуемая последовательность принадлежит
виду Onchidoris muricata .
Только одна находка из данного вида имеет 100%-совпадение с исследуемой
последовательностью. Вариабельность среди находок с %Identity выше 97% можно
об'яснить полиморфизмами, ведущими к синонимичным заменам в белке, что видно
из выравниваний, если включить трансляцию и держать в уме то, что должна быть
использована table 5 генетического кода (митохондриальный беспозвоночных)
(BLAST неправильно транслирует запрос).
Поскольку данные находки кодируют одну и ту же последовательность
белка, что и запрос, (а именно белковая последовательность для белок-кодирующих
генов имеет эволюционное значение) можно считать эти находки совпадающими.
Однако среди находок, принадлежащих виду Onchidoris muricata, имеется
одна с относительно низким процентом совпадения:

В её трансляции наблюдается 3 аминокислотные замены.
Это может означать, что существует значительная внутривидовая
вариабельность и данный участок гена cox1 недостаточно консервативен для
баркодирования видов (а может и родов, и даже семейств), а 100%-сходство
для последовательностей ДНК в одной из находок является конвергентным.
Для проверки этой гипотезы было отобрано 39 последовательностей находок,
принадлежащих разным родам. По посторенному для их трансляций (полученных с
помощью программы пакета EMBOSS transeq)
выравниванию
видно, что данный участок
гена обычно консервативен на уровне видов (исключение Knoutsodonta depressa:
имеет 1 замену Val на Ile), а иногда и на уровне родов (для рода Acanthodoris),
что не подтверждает гипотезу о возможной сильной вариабельности данного
участка гена в пределах одного вида.
Можно предположить, что обнаруженная "плохая" находка от O. muricata,
сильно отличающаяся от большего количества других находок для данного вида,
отнесена к этому виду ошибочно. Более того, её последовательность совпадает
с последовательностью одной из находок для Knoutsodonta depressa в
приведённом выравнивании.
Выравнивание полных
транслированных partial cds этих двух находок (полученных из записей NCBI)
имеет 100%-совпадение по представленным в обеих последовательностях участкам,
что подтверждает предположение, что "плохая" находка для O. muricata
на самом деле принадлежит K. depressa.
Теперь, отбросив сомнения о возможной вариабельности участка данного гена и
вспомнив о наличии 100%-совпадения после трансляции только с находками для
вида O. muricata, можно заключить, что исследуемая последовательность
является участком митохондриального гена cox1 гастроподы
Onchidoris muricata.
|
Сравнение результатов поиска разными вариантами нуклеотидного BLAST
Были сравнены результаты поиска для 2 запросов с помощью megablast, blastn с
настройками по умолчанию и blastn с максимально чувствительными настройками.
Чувствительные настройки означают настройки, позволяющие находить менее сходные
гомологичные последовательности, и предполагают короткую длину слов для поиска
(поэтому использовался Word size 7), высокое отношение reward/penalty
(использовались параметры 1, -1)[1] и низкий штраф за продление гэпа,
при существенном, но не самом высоком штрафе за открытие гэпа (исходя из того,
что в дальних гомологах могут присутствовать большие индели, но нежалательно
множественное открытие гэпов − оно может привести к негомологичным
находкам; оптимальными показались параметры штрафов 4, 1).
|
Сравнение для определённой
последовательности
гена cox1 O. muricata
Сначала был произведён поиск 3 вариантами нуклеотидного BLAST по роду
Onchidoris за исключением вида Onchidoris muricata, которому принадлежит
последовательность запроса.
megablast нашёл 11 хороших (E-value не выше 3e-80, покрытие не ниже 97%,
%Identity 82-88%) находок, и построил по 1 выравниванию для каждой.
blastn и чувствительный blastn показали похожие друг на друга результаты:
15 находок для гена cox1, из которых 13 имеют покрытие запроса
не менее 98%, а 2 оставшиеся имеют покрытие 77% и 25% или 29% соответственно, но
сохраняют хорошее E-value (не более 2e-21), и 12 находок для коротких (10-15 nt)
участков 16S rRNA и 18S rRNA (естественно, имеют неприличное E-value: от 0.97).
Различие было в количестве выравниваний для этих 27 находок: чувствительный
blastn сделал больше выравниваний в целом (57 против 43), в то же время сделав
меньше выравниваний для rRNA (12 против 17).
Видно, что megablast не очень хорошо справляется уже на уровне рода: не нашёл
2 хорошие находки и ещё 2 гомологов из-за того, что они представлены частичными
последовательностями. Дефолтный blastn и чувствительный blastn сравнить в таких
условиях трудно (слишком мало гомологов в базе, и они довольно близкие).
Для сравненения разных настроек blastn при поиске далёких гомологов был выполнен
поиск по группе Kamptozoa (сестринская моллюскам группа[2][3]):
| Алгоритм | megablast | blastn | blastn с чувствительными параметрами |
| Параметры запуска | Default | Word size: 7
Match/Mismatch Scores: 1, -1
Gap Costs Existence: 4 Extention: 1 |
| Число находок | 5 | 32 | 31 |
| Описание находок
|
Все 5 находок представляют собой короткий участок гена cox1
Loxosoma axisadversum, содержащий 25 nt-последовательность, совпадающую
с участком запроса (видимо, среди всех "кубковых червей" больше не нашлось
таких длинных совпадений). Несмотря на низкое покрытие (28%), сохраняется
хорошее E-value (4e-21), высокий %Identity (83%) обусловлен по большей части
критерием поиска.
|
29 находок представляют собой cox1 гены из всех 7 родов Kamptozoa, они имеют
покрытие запроса не ниже 85%, %Identity 66-75% и хорошее E-value (не более
3e-17), остальные 3 находки − 2 очень коротких участка mRNA Hox3 от
разных видов и не менее короткий участок mRNA Xlox от третьего вида, все с
плохим E-value 6.6.
|
29 всё тех же находок, но E-value для них стал ещё лучше (теперь не более
4e-38), а у одной находки (GenBank AC AY218083.1) значительно улучшилось
покрытие запроса с 89% до 98%, у многих других находок покрытие тоже несколько
улучшилось. Среди 2 негомологичных находок теперь mRNA Hox-гена labial и mRNA
рибосомального белка L7.
|
Graphic summary
Нажмите на изображение, чтобы увеличить | 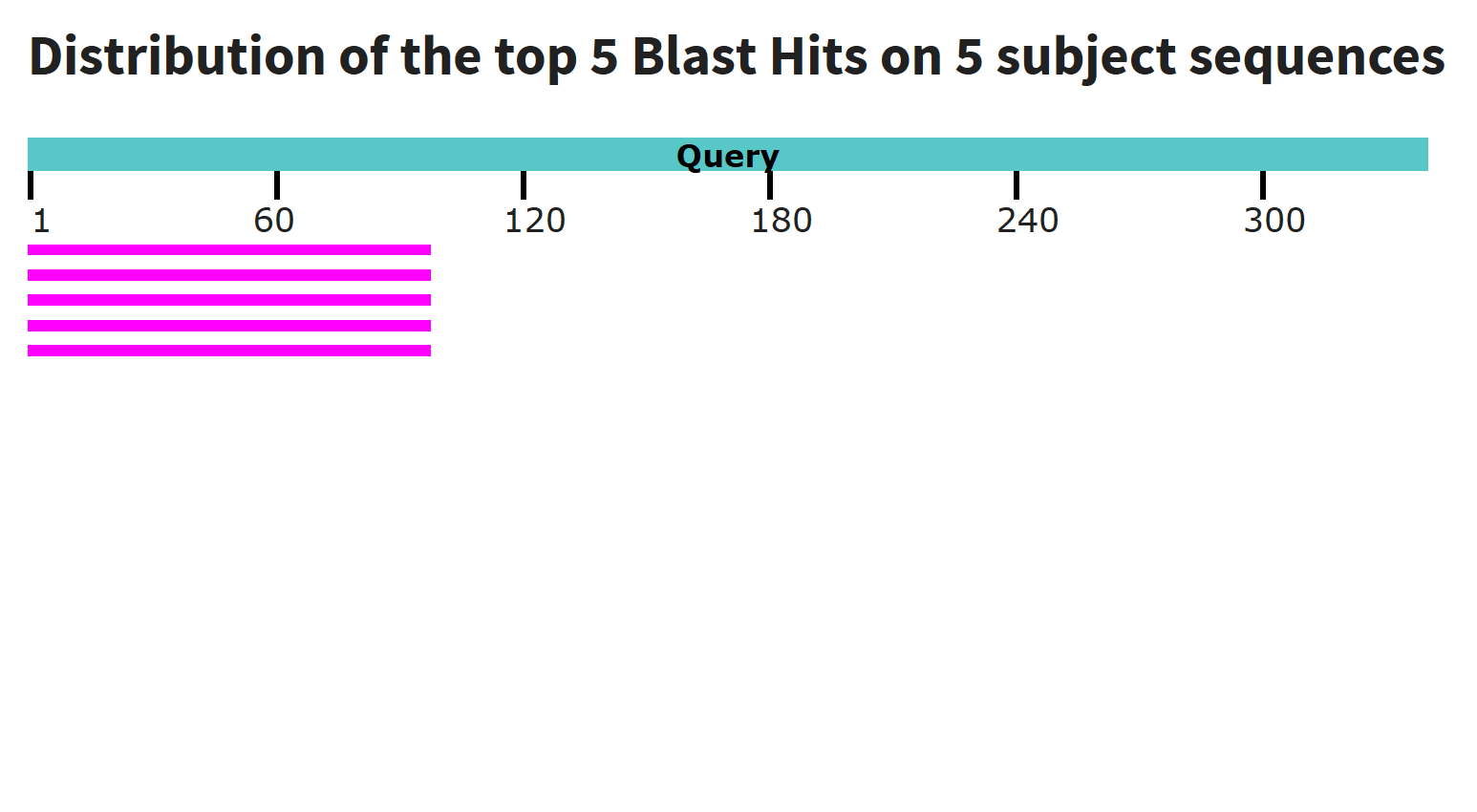 | 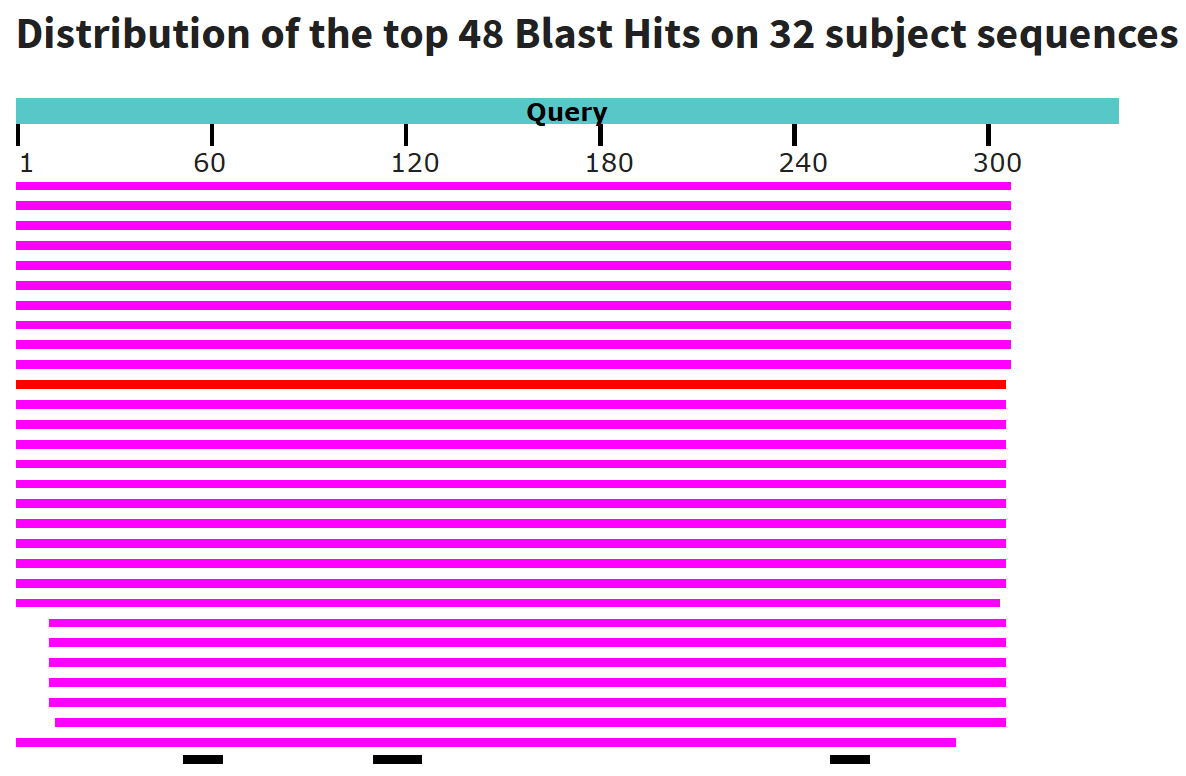 | 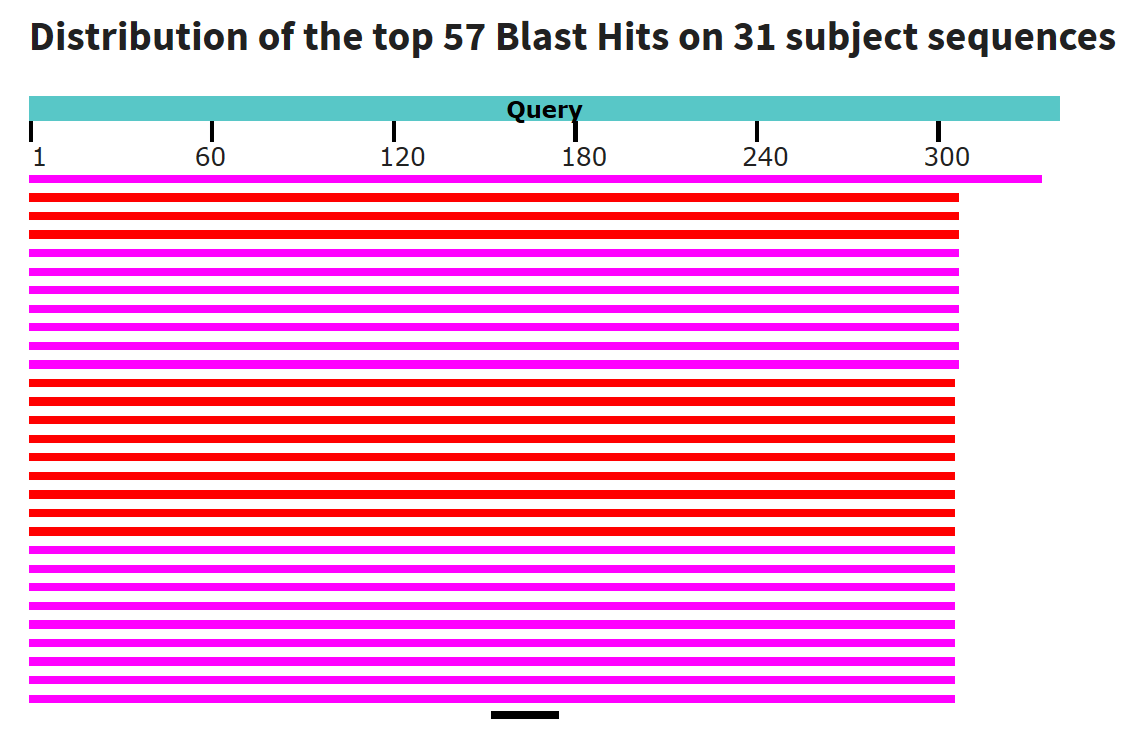 |
|
|
Сравнение для CDS
гена интегразы вируса Salmonella phage SEN1
Поиск был произведён по всей базе данных Nucleotide collection за
вычетом Proteobacteria, которые часто имеют гомологичные последовательности
вследствие горизонтального переноса генов, чем значительно увеличивают число
находок.
| Алгоритм | megablast | blastn | blastn с чувствительными параметрами |
| Параметры | Default | Default
(E-value threshold 0.001) | Word size: 7
Match/Mismatch Scores: 1, -1
Gap costs Existence: 4 Extension: 1
(E-value threshold 0.001) |
| Число находок | 4 | 30 | 59 |
| Описание находок |
Все 4 находки являются участками последовательностей записей полных геномов
фагов сальмонеллы и энтеробактерии, они имеют полное покрытие запроса и
%Identity 78% для 2 находок и 100% и 99% для 2 других. Находка со
100%-совпадением является найденной последовательностью запроса.
|
Первые 4 находки те же, что и в megablast, и имеют такие же характеристики.
Остальные находки представляют собой участки записей полных геномов или
записей сегментов ("хромосом") геномов фагов протеобактерий, так же имеется
3 записи синтетических конструкций. Хотя у этих находок наблюдается
снижение покрытия запроса до 7-26%, они имеют довольно высокий %Identity
(от 67%), большинство имеют E-value ниже 1e-4. Все находки хотя бы частично
попадают на
интегразный домен
последовательности запроса, но по сути попадают не более, чем на половину
его длины − из этого сложно сделать строгий вывод даже о гомологичности
доменов (скорее гомологичные мотивы).
|
Все находки, найденные blastn с параметрами по умолчанию, сохранились, и многие
из них значительно улучшили покрытие запроса − теперь не приходится
сомневаться в наличии у них интегразного домена, появилось 29 новых находок,
некоторые из них с хорошим покрытие запроса. Нижняя граница %Identity теперь
56%, E-value улучшилось (не выше 7e-6). Среди новых находок, в основном, участки
геномов/сегментов фагов протеобактерий, но есть и искусственные
последовательности, а также контиг некультивированной бактериии и откуда-то
участок генома Pseudomonas (протеобактерии, хотя в параметрах запуска этот
таксон исключен).
|
Graphic summary
Нажмите на изображение, чтобы увеличить | 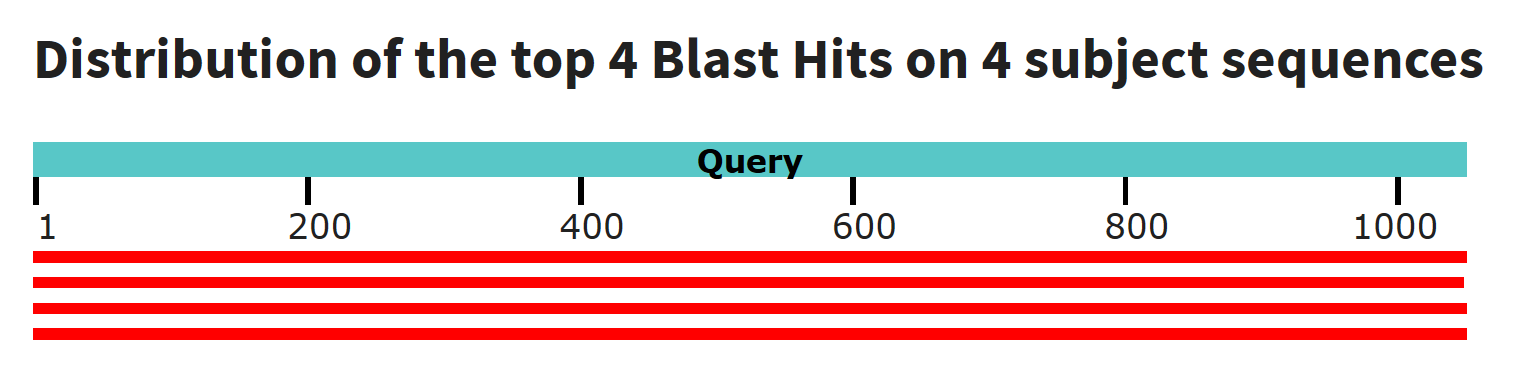 | 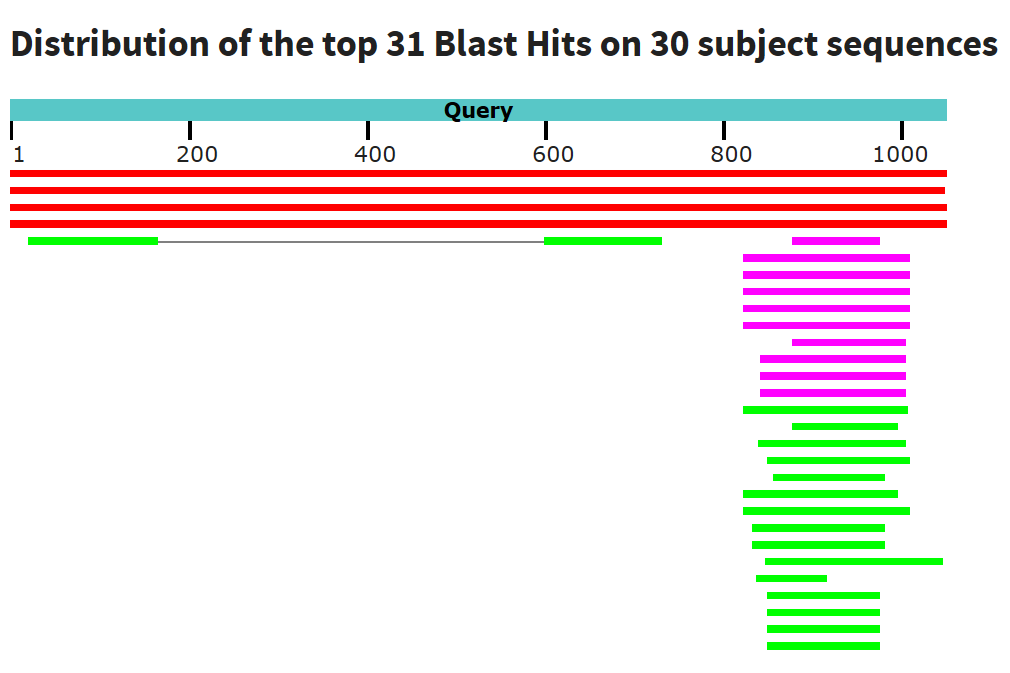 | 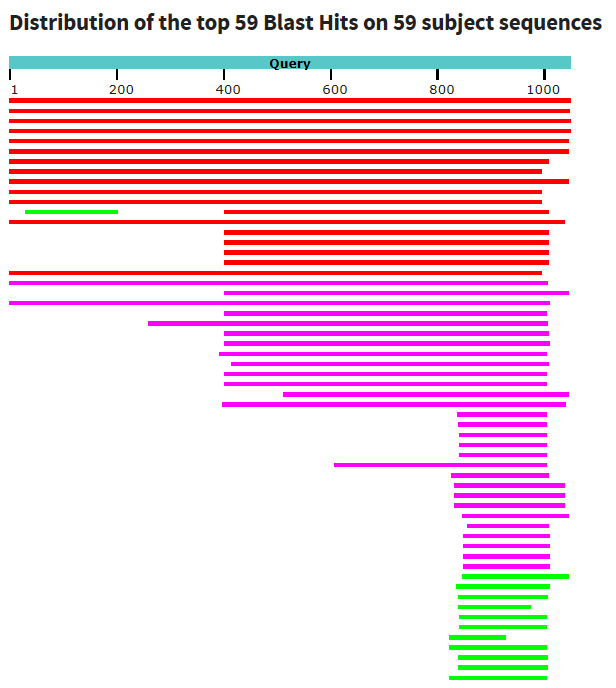 |
|
Можно заключить, что megablast подходит для быстрого поиска источника
последовательности запроса и её самых близких гомологов,
дефолтный blastn хорошо находит не самых отдалённых гомологов (лучше всего
использовать при поиске внутри одного большого таксона, к примеру отдела),
использование blastn с максимально чувствительными настройками опрадывает
себя при поиске по базе данных, содержащей в том числе отдалённые гомологи
искомой последовательности.
|
Поиск гомологов консервативных белков в неаннотированном геноме
С помощью локального BLAST на kodomo был проведён поиск 3 консервативных
белков: QDE-1, Thg1 и eRF1 − в неаннотированном геноме афелиды
Amoeboaphelidium protococcarum
Сначала на основе файла, содержащего исследуемый геном, была создана база данных
для локального BLAST:
Команда: makeblastdb -in /P/y18/term3/block2/X5.fasta -title
"Amoeboaphelidium protococcarum" -dbtype nucl -out X5
Выдача: 3 файла X5.nhr, X5.nin, X5.nsq,
вместе составляющие базу данных
Для поиска гомологов из банка UniProt были отобраны и скачены вручную
соответственные последовательности белков грибов (их UniProt ID можно найти
в выдаче BLAST), имеющие записи PDB.
В NCBI Taxonomy ядерный генетический код исследуемого вида афелид был
определён как Translation table 6.
Поиск производился программой tblastn с параметрами по умолчанию (ищет
белковый запрос в 6 возможных трансляциях нуклеотидной базы данных):
Команда: tblastn -db X5 -db_gencode 6 -query <protname>.fasta
-out <protname>.blast
Результаты поиска:
-
QDE-1 − РНК-зависимая РНК-полимераза из 2-DPBB superfamily[4][5],
широко распространённая среди эукариот (и встречающийся только у них) как
фермент, производящий вторичные siRNA.
QDE-1 происходит от фаговой ДНК-зависимой ДНК-полимеразы[5][6]
и сохраняет характерную доменнную организацию,
включающую оба DPBB-домена, вместе образующих каталитический центр, в отличии
от мультисуб'единичных полимераз, у которых менее консервативный DPBB1 находится
в cуб'единице β, а более консервативный DPBB2, несущий каталитические
остатки, находится в cуб'единице β'.
В выдаче BLAST имеется 2 практически одинаковые
находки из разных скэффолдов. Несмотря на хороший E-value, находки имеют низкий
(практически граничный) %Identity 28%. Однако исходя из соответствующей PDB
структуры (PDB ID 2J7N), можно наблюдать, что находки включают в себя оба
DPBB-домена и наибольшее число совпадений приходится как раз на каталитически
активные участки (остатки 647–807 и 914–1161[4]), в DPBB2-домене
сохранены 3 каталитических остатка Asp (D1007, D1009, и D1011), а все гэпы
приходятся на менее консервативный neck-участок. По наличию обоих DPBB-доменов
и всех 3 каталитических остатков, можно с уверенностью утверждать о наличии
у исследуемой афелиды 2 гомологов QDE-1 (причём, судя по схожести
последовательности, образовавшихся при недавней дупликации), низкое покрытие
запроса вполне ожидаемо и об'яснимо низкой консервативностью некаталитических
участков.
-
Thg1 − фермент, характерный только для эукариот, участвующий в созревании
tRNAHis, осуществляя присоединение G-1 на 5'-конец
молекулы. Thg1 входит в семейство TLP (Thg1-like proteins) полимераз,
распространённых среди бактерий и архей, включая эукариот, и осуществляющих
уникальную 3'-5'-полимеразную реакцию для репарации и редактирования
5'-конца различных tRNA; семейство TLP, в свою очередь, входит в Klenow
superfamily[7].
Эукариоты могут иметь непосредственно Thg1 и ферменты из группы эукариотических
TLP; Thg1 отличается наличием характерной последовательности HINNLYN (мотив 2)
после фрагмента Клёнова и вариантом последовательности активного сайта в петле
сразу перед третьим β-тяжом фрагмента Клёнова (мотив 1) SDE(Y/F)SF
(вместо SDEI(T/N)(M/L) ) у других TLP)[7].
В выдаче BLAST имеется 2 похожих друг на друга
находки, они обладают хорошим %Identity (в среднем около 45%) и имеют практически
полное покрытие (если учитывать в каждой находке оба выравнивания). Обе находки
имеют характерные для Thg1 мотивы 1 и 2, также сохранён и другой активный
центр (D29)[8], 3 большие инсерции (включая участок со смещением
рамки считывания) при тщательном рассмотрении оказались
первым,
вторым и
третьим интронами (выделены границы
интронов, сплайсинг-сайты и предполагаемые branching adenines),
индели в первом или третьем интроне приводят к сдвигу рамки в первой
или второй находке соответственно.
Таким образом, данный вид афелид имеет 2 гена фермента Thg1.
-
eRF1 − class 1 release factor, связывается с любыми стоп-кодонами в
A-сайте рибосомы для последующего освобождения транслированной полипептидной
цепи и терминации трансляции.
Из выдачи BLAST видно, что геном исследуемой
афелиды содержит 2 явных гомолога (высокий %Identity и почти полное покрытие).
Так же имеется странная короткая находка, сильно похожая на N-концевой участок
eRF1; хороший E-value (6e-9) не позволяет подозревать здесь случайную находку
− видимо, действительно гомологичный участок. Исходя из эволюционной
истории трансляционных фактров[9], не приходится ожидать, что эта
короткая находка − участок какого-то другого фактора трансляции.
Но тогда что это такое? Попробуем узнать в следующем задании.
|
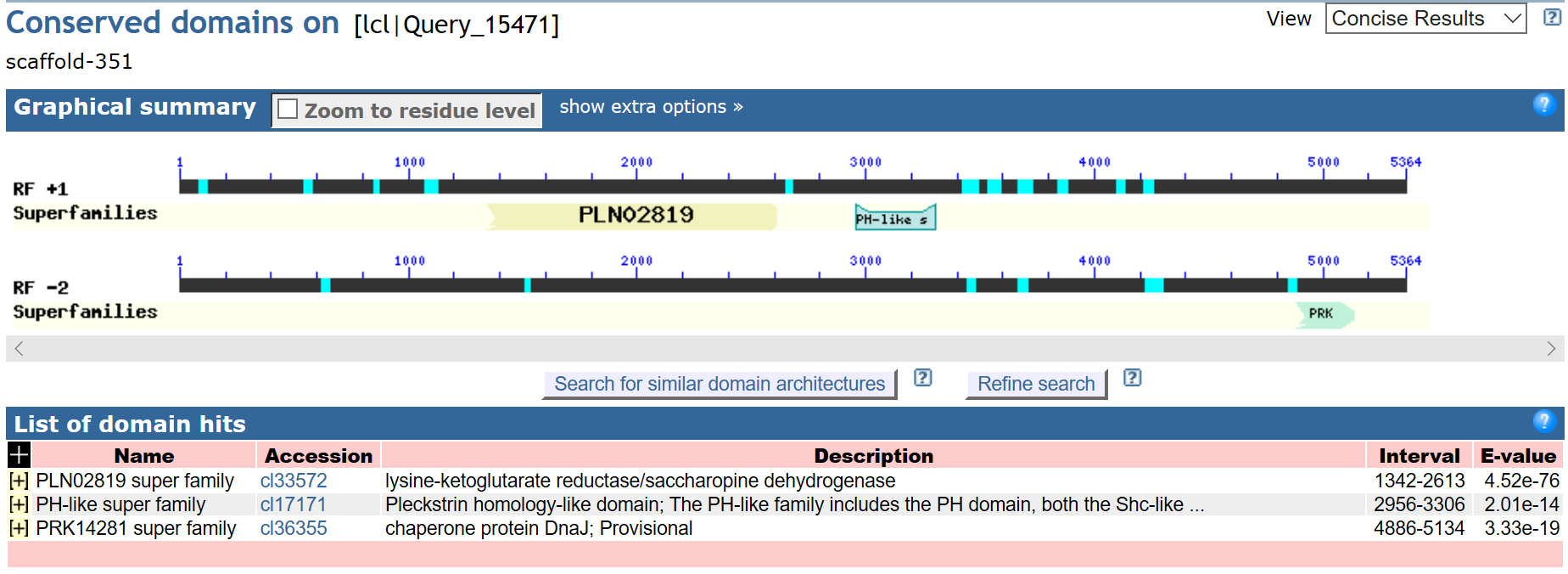
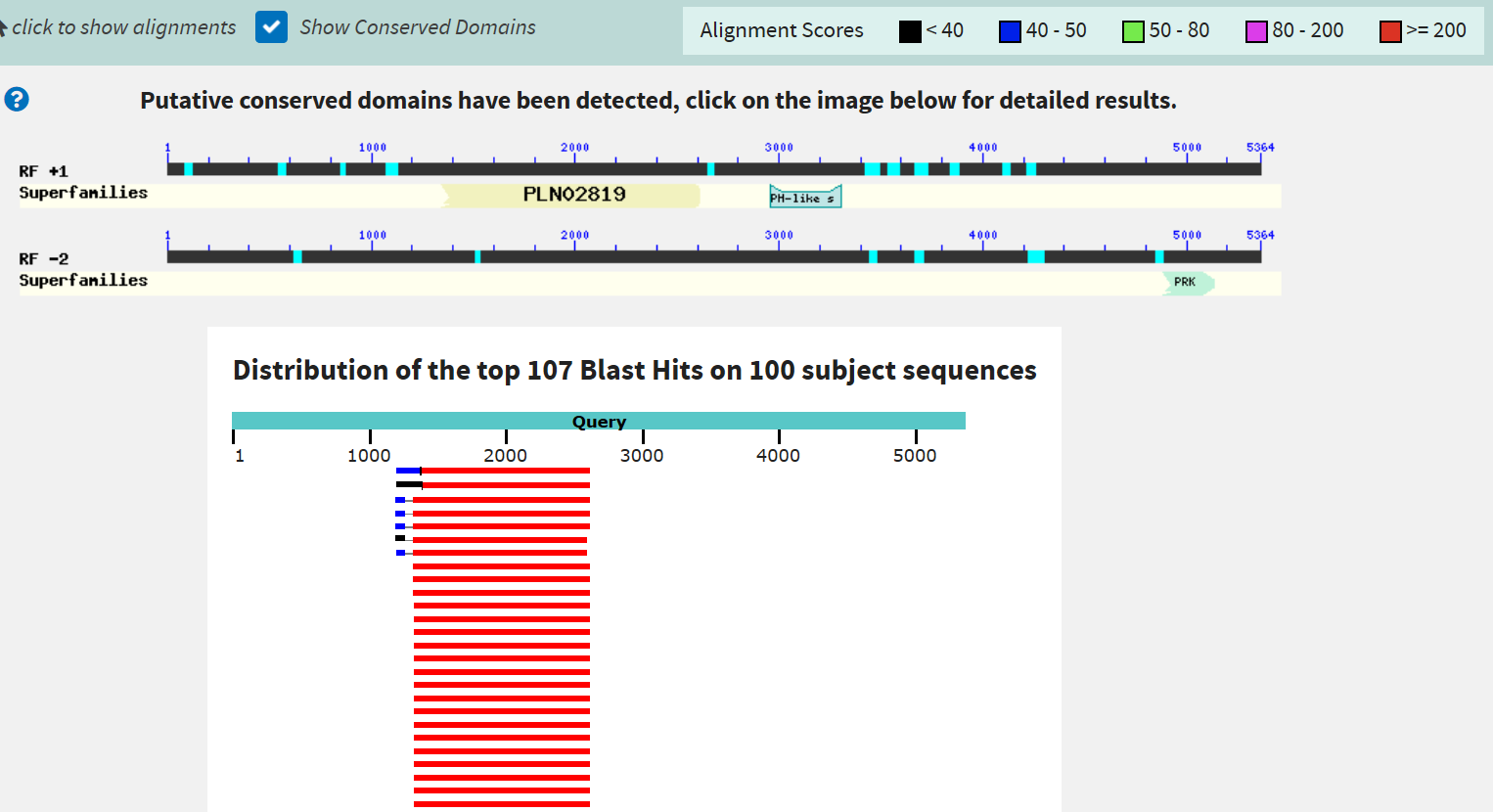
Поиск белок-кодирующих генов в скэффолде
Для определения белок-кодирующих генов в скэффолде 351 (содержащем N-концевой
участок eRF1) из неаннотированной сборки генома
Amoeboaphelidium protococcarum на сайте NCBI с помощью
blastx с параметрами поиска по умолчанию был произведён поиск
последовательности этого скэффолда (транслированной в 6 рамках по Translation
table 6) по базе Reference proteins среди организмов группы
Opisthokonta за исключением Metazoa.
blastx смог определить 3 гена в
скэффолде. Наиболее поддержанным из них можно считать
сахаропиндегидрогеназу, к ней относятся
первые 100 находок, они
все имеют низкий E-value (не выше 3e-157) и хороший %Identity
(от 55% до 61%).
Из выравниваний и последовательности в записи любой из находок видно, что
запрос имеет практически полное покрытие находок, т.е. кодирующая
последовательность определённого гена консервативна по всей длине. Можно
попытаться определить координаты CDS.
На графической выдаче BLAST видно, что
некоторые находки имеют гомологичный отдельно стоящий N-концевой участок,
однако он закодирован в другой рамке считывания.
Такая ситуация может возникать в 4 случаях:
имеется сдвигающий рамку индель в интроне, перед нами псевдоген, имеется
ribosome slippery sequence, ошибка сборки, − первый случай кажется
наиболее правдоподобным.
В предположительном начале первого CDS (определёно по находке для
Spizellomyces sp.) был определён старт-кодон и найден завершающий его
интрон, сдвигающий рамку считывания:
последовательность начала гена
В предположительном конце второго CDS был найден стоп-кодон:
последовательность конца гена
Таким образом, в скэффолде 351 определён ген сахаропиндегидрогеназы −
фермента, участвующего в метаболизме лизина, − с CDS
1179..1266, 1328..2619.
А что же с находкой из начала этого скэффолда, похожей на N-концевой
участок eRF1? Дефолтный blastx не обнаружил ничего похожего среди первых
находок, а также не нашёл каких-либо характерных доменов в соответствующем
участке скэффолда. Поиск с якорным словом длины 2 участка скэффолда 1..1100
по той же базе даннных и тем же организмам выдаёт множество находок, являющихся
факторами терминации eRF1 или похожими putative proteins, все которые попадают
на один участок и всё ещё имеют небольшое покрытие скэффолдом (10-12%). E-value
у этих находок остаётся приличным (не более 3e-07 для 100 находок).
Сложно понять, что это такое. Скорее всего данная последовательность
образовалась при транспозиции участка последовательности CDS гена eRF1, причём,
судя по сохранению последовательности при предполагаемом отсутствии функции,
эта транспозиция произошла недавно (скорее всего на уровне рода или вида).
|
Ссылки
-
BLAST topics
-
Levy, J. (2019) Computational Methods to Resolve Deep Species Phylogenies
-
Haszprunar, G., Wanninger, A. (2008) On the fine structure of the creeping
larva of Loxosomella murmanica: additional evidence for a clade of Kamptozoa
(Entoprocta) and Mollusca
-
Salgado, P. et al. (2006) The Structure of an RNAi Polymerase Links RNA
Silencing and Transcription
-
Sauguet, L. (2019) The Extended ‘Two-Barrel’ Polymerases Superfamily:
Structure, Function and Evolution
-
Drobysheva, A. et al. (2020) Structure and function of
virion RNA polymerase of crAss-like phage
-
Jackman, J. et al. (2012) Doing it in reverse: 3'-to-5' polymerization by the
Thg1 superfamily
-
Hyde, S. et al. (2010) tRNAHis guanylyltransferase (THG1), a unique
3′-5′ nucleotidyl transferase, shares unexpected structural homology
with canonical 5′-3′ DNA polymerases
-
Inagaki, Y. (2000) Evolution of the Eukaryotic Translation Termination System:
Origins of Release Factors
|
|
|
{kind=link}
{kind=link}