|
Поиск SD-последовательностей в геноме P. abyssi
I. Сначала был проведён тестовый поиск обеими простроенными PWM в 500
upstream-последовательностях длиной 20 nt из CDS, отобранных случайным образом
из набора "хороших" белок-кодирующих генов (см. выше):
-
Из набора "хороших" CDS были случайно отобраны 500 записей, для них были
определены координаты upstream-последовательнотей длиной 20 nt с учётом
ориентации кодирующей цепи;
-
Из набора определённых на предыдущем шаге координат и fasta файла сборки
хромосомы скриптом fragments2fasta.py был получен fasta файл с
отобранными 500 upstream-последовательностями;
-
Поиск производился с последовательным применением обеих построенных PWM
программой FIMO только на данных цепях сначала с
p-value cutoff 0.001, затем – с p-value cutoff 0.01.
При поиске мотива GGTGA (далее: короткого мотива) в 500
upstream-последовательностях программой FIMO с p-value cutoff 0.001
было найдено всего
132 сайта данного мотива,
26.4% от числа входных последовательностей, что намного меньше
ожидаемого числа CDS с SD-зависмой инициацией трансляции у взятой
археи[3]. Все найденные последовательности совпадают с консенсусом
мотива.
При поиске FIMO с p-value cutoff 0.01 нашлось 450 сайтов данного мотива,
последовательности сайтов стали более разнообразными. Дальнейший анализ
проводился для этого набора сайтов:
Несмотря на разумный порог p-value, не все находки в наборе действительно
значимы (multiplicity problem), поэтому решено было отобрать часть из них на
основе q-values. При FDR = 0.05 было слишком много потерь, поэтому был принят
FDR = 0.1.
Среди найденных сайтов были те, которые попадали дважды на одну
upstream-последовательность, что биологически бессмысленно. Поэтому для каждой
последовательности с множеством найденных сайтов была отобрана первая в таблице
находка с наименьшим p-value (та же, что и с наибольшим score).
Была составлена таблица
с названиями и координатами генов, в которых был найден
сайт с мотивом, и указанным расстоянием от сайта до старт-кодона.
Таким образом, было найдено 344 сайта мотива, то есть 344 из 500 взятых CDS
(68,8%) несут SD последовательность. Из распределения последовательностей
найденных сайтов видно, что большинство соответствует консенсусу или близко
к нему:
GGTGA 131
GGAGG 79
GGTGG 50
GGGGA 29
GGAGA 24
GGTGT 20
GGTGC 7
GGCGA 4
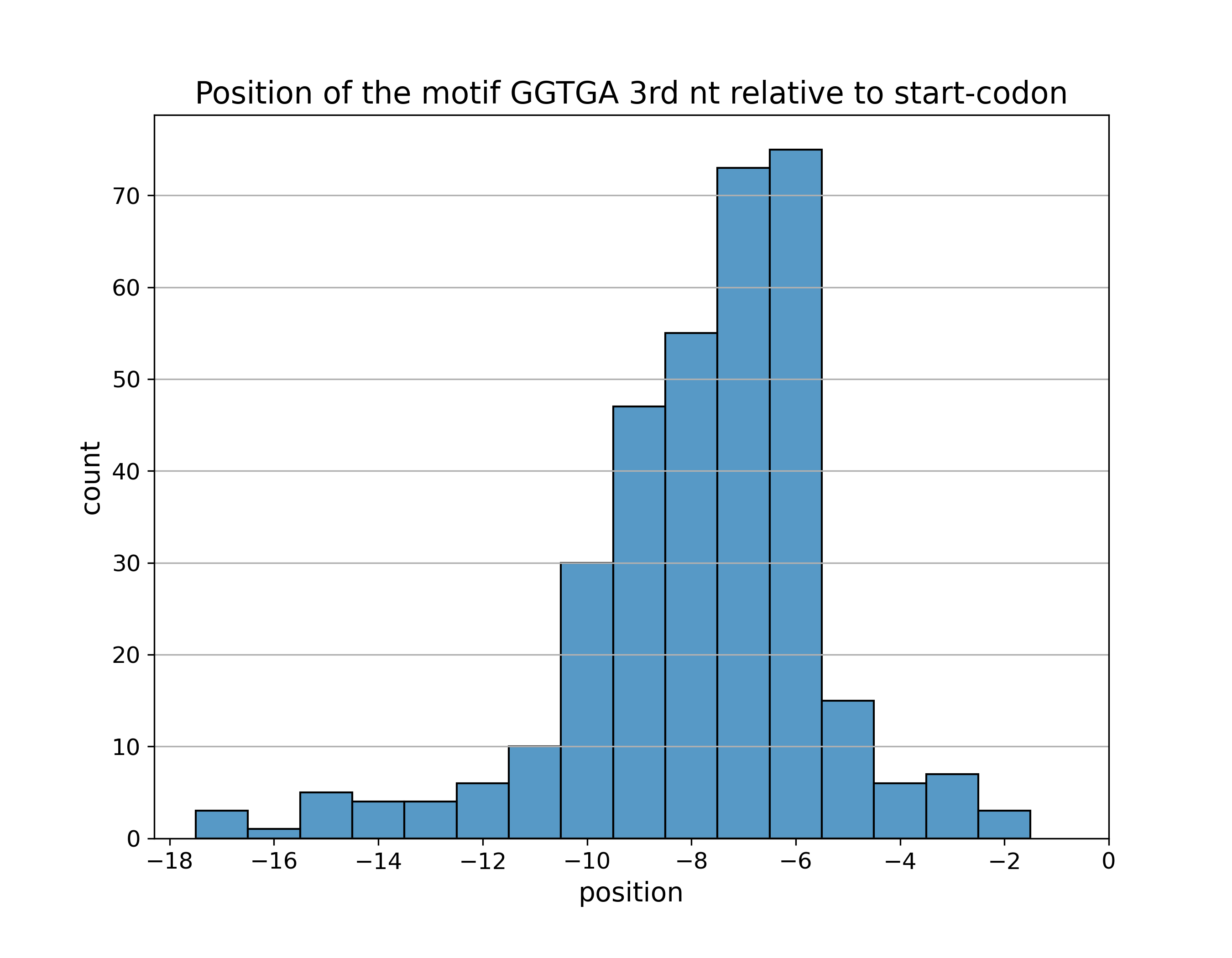
Для отобранных сайтов мотива построена гистограмма расстояний от
III нуклеотида поледовательности мотива до старт-кодона
(или точнее, бар-плот координат этого нуклеотида):
Для отобранных последовательностей сайтов мотива построено LOGO мотива
(сервис WebLogo3, параметр no adjustment for composition):
При поиске мотива GGAGGTGAK (далее: длинного мотива)
в 500 upstream-последовательностях программой FIMO с
p-value cutoff 0.001 было найдено всего
169 сайтов данного мотива,
32.8% от числа входных последовательностей, что снова мало. Находки имели
различные последовательности.
При поиске FIMO с p-value cutoff 0.01 нашлось 555 сайтов данного мотива.
Дальнейший анализ проводился для этого набора сайтов:
При отборе наход по q-value, как и в прошлый раз был взят FDR = 0.1. Для
избавления от множественных сайтов на upstream-последовательностях, из каждой
группы таких сайтов снова выбирался первый в таблице сайт с наименьшим p-value.
Была составлена таблица
с названиями и координатами генов, в которых был найден
сайт с мотивом, и указанным расстоянием от сайта до старт-кодона.
Таким образом, было найдено 329 сайтов мотива, то есть 329 из 500 взятых CDS
(65.8%) несут SD последовательность. Разнообразие найденных
последоватеьностей велико (не более 8 сайтов один тип последовательности
мотива). Было оценено количество характерных для SD мотива
последовательностей в ожидаемо наиболее консервативном участке найденных сайтов
(4..8 nt), а также количество сайтов, где в этом участке присутствует
нехарктерный для SD мотива цитозин:
include GGTGA 105
include GGTGA or GGAGG or GGTGG or GGGGA 174
include C 43
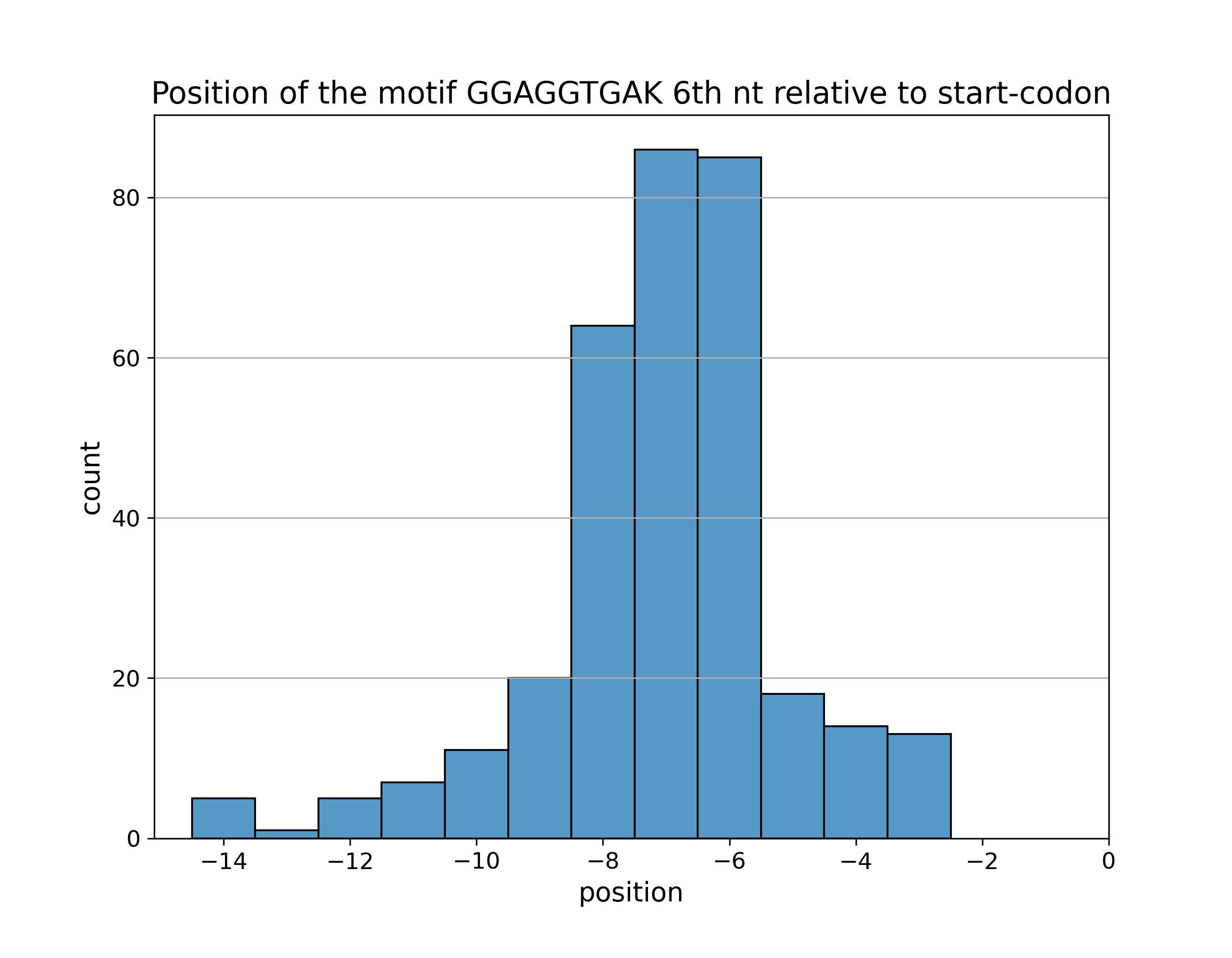
Для отобранных сайтов мотива построена гистограмма расстояний от
VI нуклеотида последовательности мотива до старт-кодона:
Для отобранных последовательностей сайтов мотива построено LOGO мотива
(сервис WebLogo3, параметр no adjustment for composition):
Несмотря на хорошее сходство с последовательностью 3'-концевого участка
16S rRNA, длинная PWM находит в upstream-последовательностях 500 отобранных
"хороших" CDS несколько меньше достоверных сайтов мотива, и значительно большая
часть найденных сайтов имеет нехарактерные для искомого мотива
последовательности по сравнению с поиском короткой PWM.
Для дальнейшего поиска мотива SD во всех белок-кодирующих генах хромосомы
археи будет использоваться короткая PWM.
II. Затем был произведён поиск SD мотива, заданного короткой (более
строгой) PWM, в upstream-последовательностях всех аннотированных CDS
исследуемой сборки:
-
Для всех 1783 CDS сборки, координаты которых были получены ранее с помощью
скрипта features2CDSs.py (см. выше), с учётом ориентации кодирующей
цепи были определены координаты 20 nt upstream - 15 nt downstream
последовательностей для поиска SD мотива, downstream участок взят для возможных
случаев неверной аннотации старт-кодона;
-
Из набора определённых на предыдущем шаге координат и fasta файла сборки
хромосомы скриптом fragments2fasta.py был получен fasta файл с
отобранными 1783 upstream-downstream последовательностями;
-
Поиск производился применением короткой PWM программой FIMO только на
данных цепях с p-value cutoff 0.001 и затем p-value cutoff 0.01.
При поиске короткого мотива с p-value cutoff 0.001 находилось
всего 587 сайтов, что составляет
32.9% от числа входных последовательностей. Последовательности всех найденных
сайтов совпадали с консенсусом искомого мотива.
При поиске с p-value cutoff 0.01 нашлось 2032 сайта данного мотива. Дальнейший
анализ проводился для этого набора сайтов:
Для отбора по q-values был взят FDR = 0.1. Для избавления от множественных
сайтов на upstream-последовательностях, из каждой группы таких сайтов
выбирался первый в таблице сайт с наименьшим p-value.
Была составлена таблица с названиями
и координатами генов, в которых был найден сайт с мотивом, и указанным
расстояние от последнего нуклеотида сайта до аннотированного старт-кодона.
Таким образом, было найдено 1161 сайт мотива, то есть для 1161 из 1783 (65.1%)
CDS хромосомы были обнаружены сайты SD мотива, что однако всё ещё меньше
предыдущих оценок[3].
Из распределения последовательностей найденных сайтов видно, что они все
вполне похожи на последовательность SD:
GGTGA 560
GGAGG 247
GGTGG 240
GGAGA 114
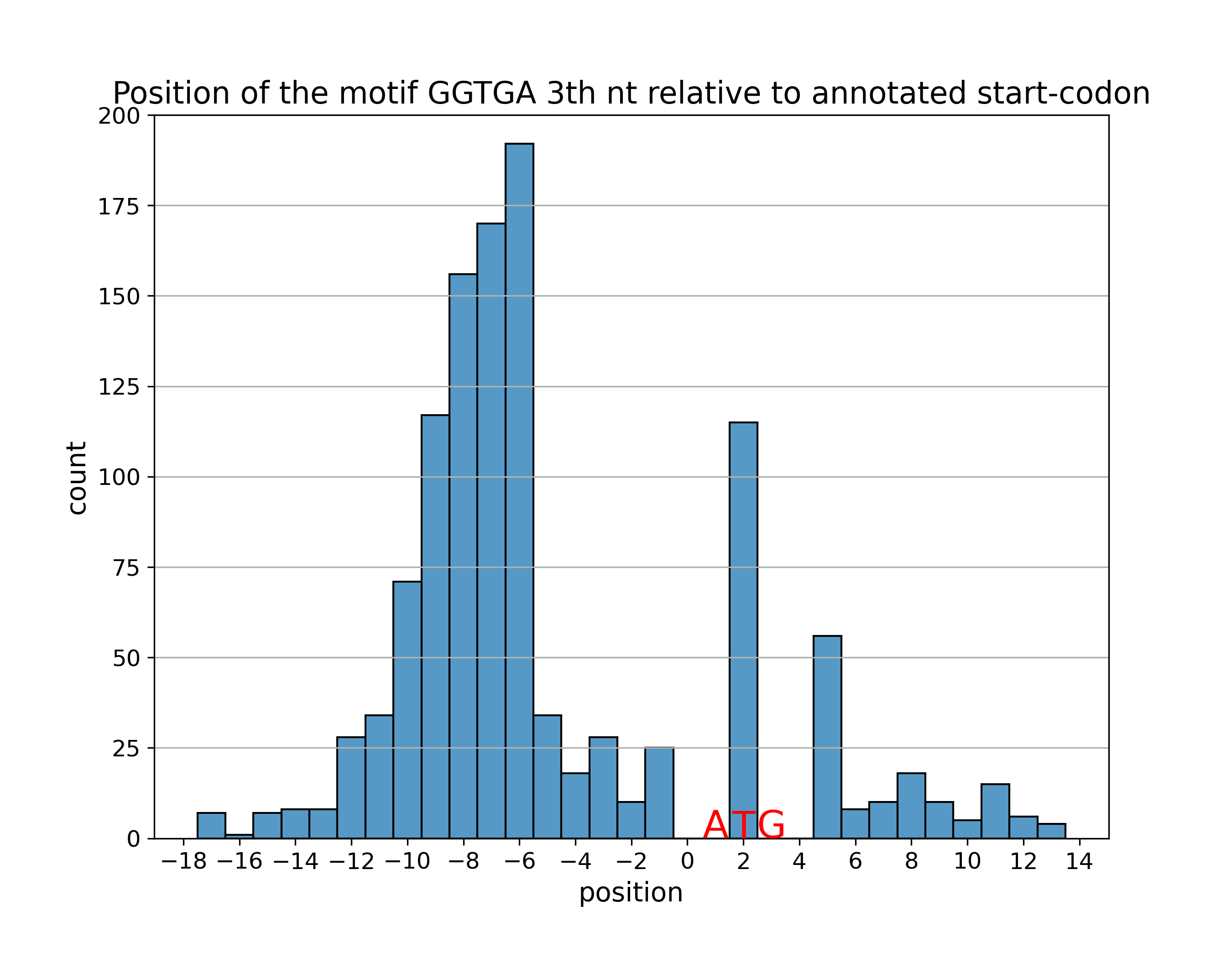
Для отобранных сайтов мотива построена гистограмма расстояний от
III нуклеотида поледовательности мотива до аннотированного старт-кодона
(или точнее, бар-плот координат этого нуклеотида):
Как видно из гистограммы расстояний и таблицы найденных сайтов выше
значительная часть сайтов была найдена downstream к аннотированному
старт-кодону или "внутри" старт-кодона. Биологически это невозможно,
объяснить такое состояние дел можно либо ложной находкой сайта SD,
либо неверно аннотированным старт-кодоном.
Первое объяснение вполне вероятно: в силу нестрогого порога FDR до 10% наход
могут быть ложноположительными. Однако "заскоков" за старт-кодон больше, чем
10% находок (23.4%) и распределение последовательностей этих находок вполне
похоже на общее распределение (нет преобладания наименее характерных
последовательностей):
GGTGA 130
GGTGG 57
GGAGA 51
GGAGG 34
Поэтому второе объяснение тоже заслуживает внимания, учитывая, что среди CDS
сборки много записей с плохо аннотированными названиями/без названия. Однако
χ2-тест на независимость переменных не показал связи между
качеством названия гена и положением найденного SD сайта:
Observed:
bad name False True
wrong start
False 632 257
True 183 89
chi2-test p-value = 0.259
Таким образом, общую причину не удаётся выявить простыми методами.
В дальнейшем, при построении LOGO мотива считалось, что все найденные сайты
определены верно, т.е. подразумевалось, что причина всегда в неверно
аннотированном старт-кодоне (что, даже если неверно, вряд ли сильно скажется
на LOGO из-за сходного распределения последовательностей).
Для отобранных последовательностей сайтов мотива построено LOGO мотива
(сервис WebLogo3, параметр no adjustment for composition):
|