Учебный сайт Шиндяпиной А.В.
Пространственное выравнивание и совмещение
PyMol команда align
С помощью команды align в PyMol сделала совмещение цепи А из белков 1HY0 и 1I0A по каждому
из доменов (предсказаны CATH). Для обоих белков границы доменов совпадают:
1: 27-123, 195-234
2: 124-194, 235-364, 437-463
3: 365-436
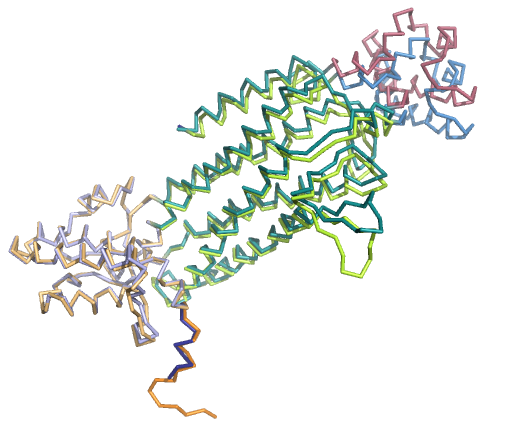
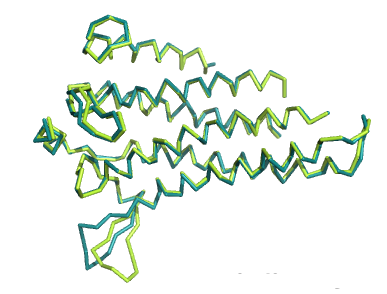
Ниже представлены изображения по всем трем совмещениям и их значения RMSD (rms в PyMol):
1. RMSD=0.371

2. RMSD=0.409
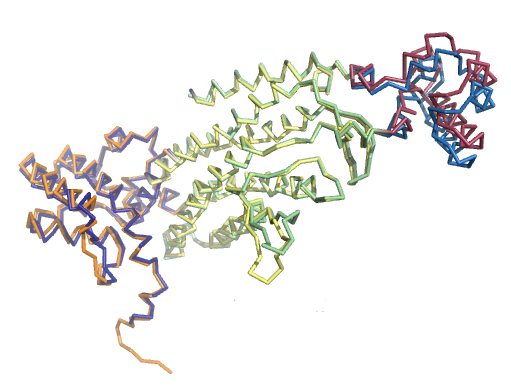
3. RMSD=0.527

Как видно, при совмещении по 1-м и по 3-м доменам остальные части цепей сильно расходятся.
В случае всех трех совмещений по доменам RMSD очень хороший (<0.6). Из чего можно сделать вывод, что при задаче совмещения
структур белков чаще будет правильнее смотреть совмещение не по всей их структуре, а по
более консервативным, доменным участкам.
Команды pair_fit, align и super
Для того, чтобы воспользоваться командой pair_fit для начала с помощью сервиса Geometrical core
определила геометрическое ядро структур цепи A 1AKH и 1W0T:
| Pos. |
1AKH_A |
1W0T_A |
| 11 |
ALA83 |
LYS389 |
| 12 |
PHE84 |
ASN390 |
| 13 |
LEU85 |
LEU391 |
| 15 |
GLU87 |
SER393 |
| 28 |
LYS100 |
TRP403 |
| 29 |
GLU101 |
SER404 |
| 30 |
GLU102 |
LYS405 |
| 31 |
VAL103 |
ILE406 |
| 41 |
THR110 |
THR416 |
| 42 |
PRO111 |
SER417 |
| 43 |
LEU112 |
VAL418 |
| 44 |
GLN113 |
MET419 |
| 45 |
VAL114 |
LEU420 |
| 46 |
ARG115 |
LYS421 |
| 47 |
VAL116 |
ASP422 |
| 48 |
TRP117 |
ARG423 |
| 49 |
PHE118 |
TRP424 |
| 50 |
ILE119 |
ARG425 |
| 51 |
ASN120 |
THR426 |
| 52 |
LYS121 |
MET427 |
| 53 |
ARG122 |
LYS428 |
| 54 |
MET123 |
LYS429 |
| 55 |
ARG124 |
LEU430 |
Выделив эти два множества (только ca атомы):
select core1, 1akh and chain a and name ca and (resi 83-85+87+100-103+110-124)
select core2, 1w0t and chain a and name ca and (resi 389-391+393+403-406+416-430)
сделала пространственное выравнивание командой pair_fit core1, core2:
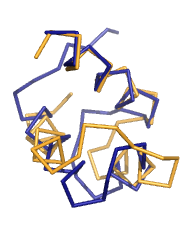
Как видно, выравнивание получилось довольно хорошое, RMSD=0.865.
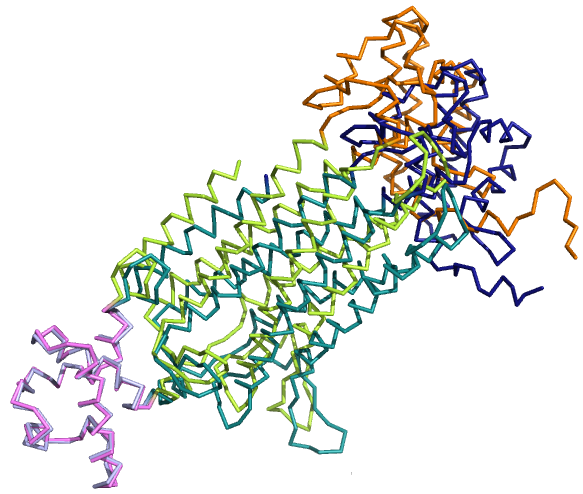
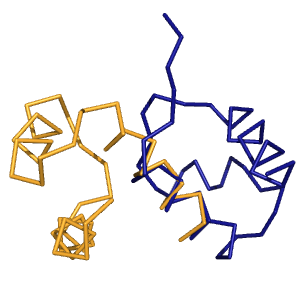
Выравнивание, полученное командой align для полных цепей, явно не имеет никакого биологичекого смысла,
т.к. выравнилось только 69 атомов (и то для них RMSD=1.340):
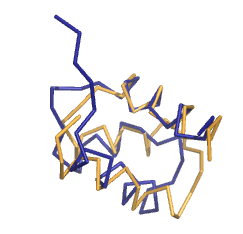
Выравнивание командой super намного более смысловое, чем предыдущее, но RMSD у него 2.186, меньше чем в случае pair_fit, т.е. оно хуже.
©, "ООО Шиндяпина 2008"