| Главная | Семестры | Обо мне | Ссылки |
|---|
Анализ качества и очистка чтений
Анализ качества чтений
Программа FastQC предназначена для чтения и анализа файлов в формате fastq.
На выдаче можно получить отчет в формате html. Команды:
gunzip Ath_tae_CTTGTA_L003_R1_004.fastq.gz
fastqc Ath_tae_CTTGTA_L003_R1_004.fastq
Отчет: открыть.
На графике Per sequence quality scores видим, что максимальное количество ридов имеет качество 38 (вероятность ошибки между 1/1000 и 1/10000).
Будем далее использовать формат fastq phred33.
Очистка чтений
Задача: удалить последовательности адаптеров, отрезать с конца каждого рида нуклеотиды с качеством ниже 20, оставить только риды длиной не меньше 50 нуклеотидов.
Для этого используется программа Trimmomatic.
Все файлы с примерами адаптеров были объединены в файл adapters.fasta.
Команды:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 Ath_tae_CTTGTA_L003_R1_004.fastq outfile1.fastq ILLUMINACLIP:adapters.fasta:2:7:7
bljava -jar /usr/share/java/trimmomatic.jar SE outfile1.fastq outfile2.fastq TRAILING:20
java -jar /usr/share/java/trimmomatic.jar SE outfile2.fastq out.fastq MINLEN:50
fastqc out.fastq
Отчет FastQC: открыть.
Сравнение ридов до и после обработки Trimmomatic
Из 4000000 ридов осталось 3858095, то есть было удалено 141905 ридов.
Per base sequence quality. См. рис. 1-2. Так как были удалены хвостовые нуклеотиды с качеством ниже 20, среднее значение качества нуклеотидов выросло. Особенно это заметно для концевых ридов (начиная с 90-й позиции).
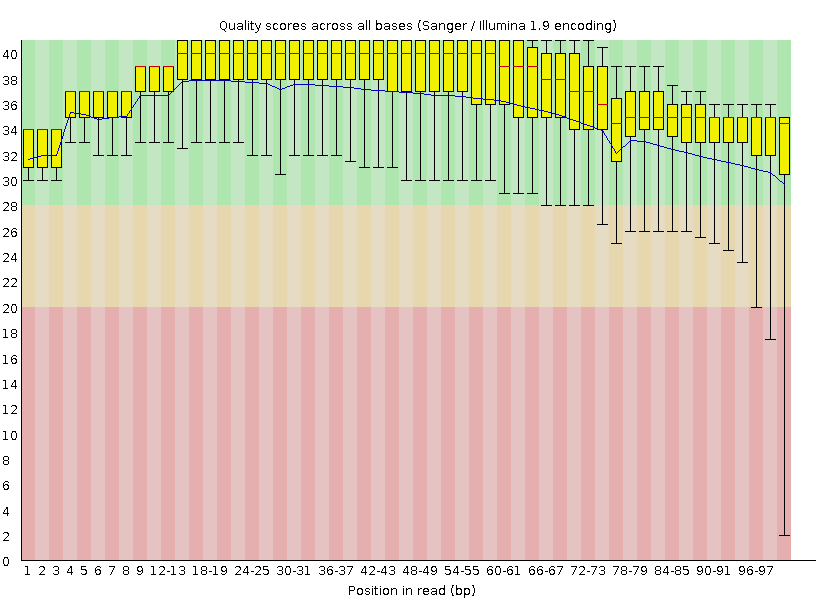
Рис. 1. Per base sequence quality до обработки Trimmomatic. Получено с помощью FastQC
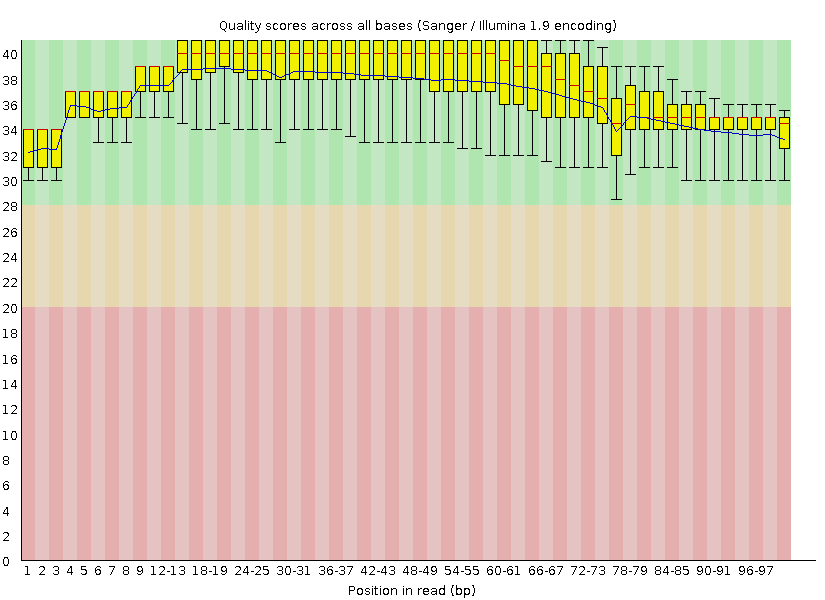
Рис. 2. Per base sequence quality после обработки Trimmomatic. Получено с помощью FastQC
Per base sequence content. См. рис. 3-4. Визуально графики не различаются. Из раздела Basic Statistics узнаем, что процент GC-пар не изменился и составляет 35%.
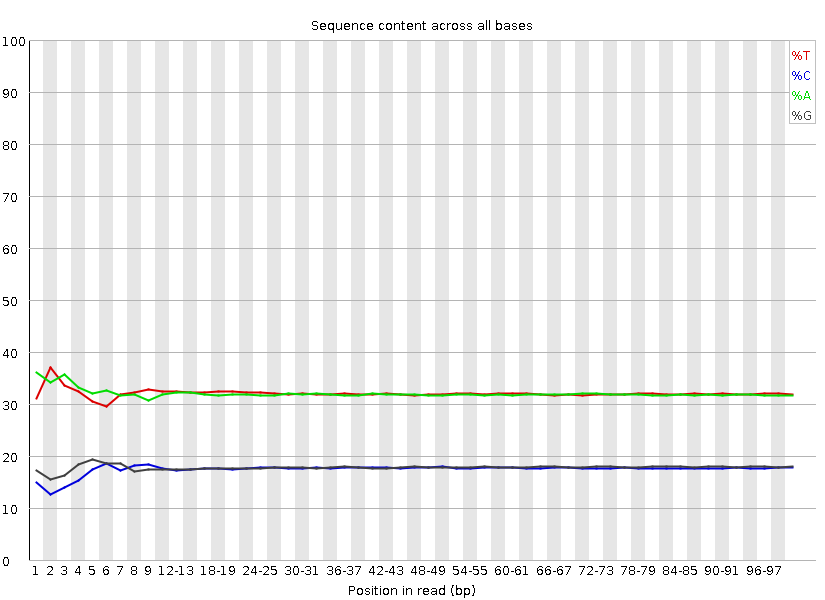
Рис. 3. Per base sequence content до обработки Trimmomatic. Получено с помощью FastQC
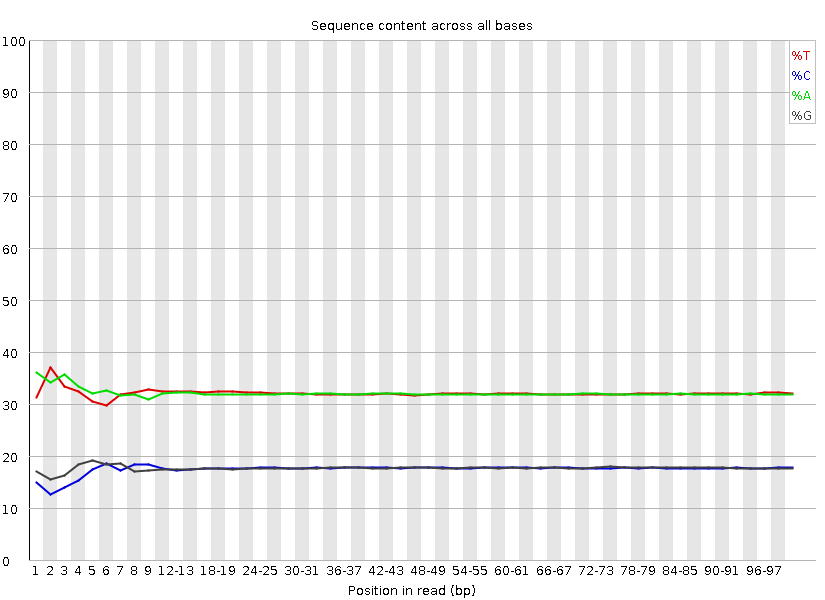
Рис. 4. Per base sequence content после обработки Trimmomatic. Получено с помощью FastQC
Sequence length distribution. См. рис. 5-6. Изначально длина всех ридов была равна 101. После обрезки низкокачественных хвостовых нуклеотидов и удаления ридов длины менее 50 мы видим, что появляются более короткие риды. Но их доля относительно общего количества невелика, и большая часть ридов все так же имеет длину 100-101 нуклеотид. Это говорит о хорошем качестве исходных ридов.
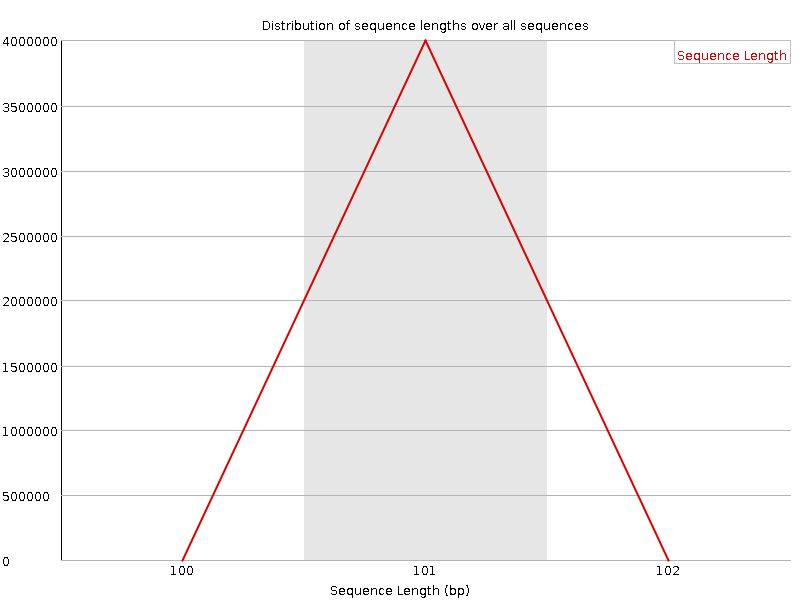
Рис. 5. Sequence length distribution до обработки Trimmomatic. Получено с помощью FastQC
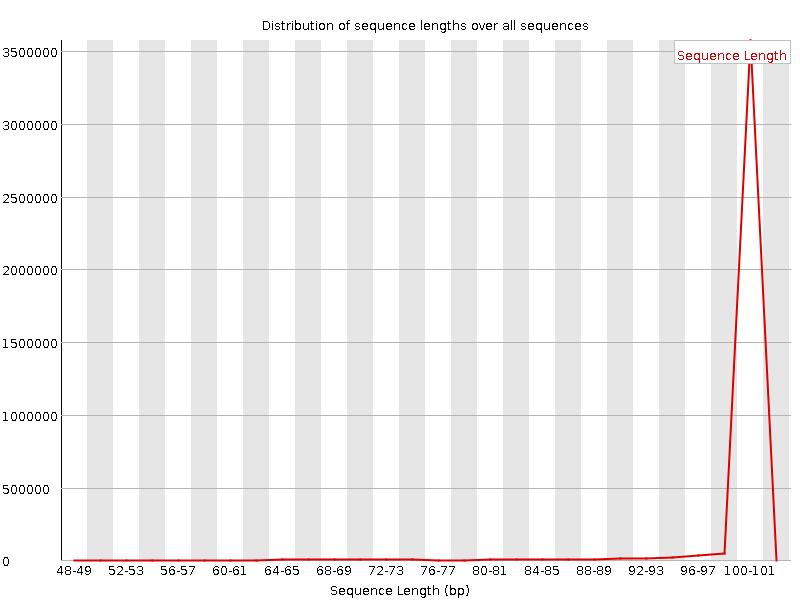
Рис. 6. Sequence length distribution после обработки Trimmomatic. Получено с помощью FastQC