При помощи BLAST очень удобно проводить поиск последовательностей, гомологичных данной по различным базам данных.
Также BLAST автоматически создает парное выравниванеи найденной последовательности с запрашиваемоей.
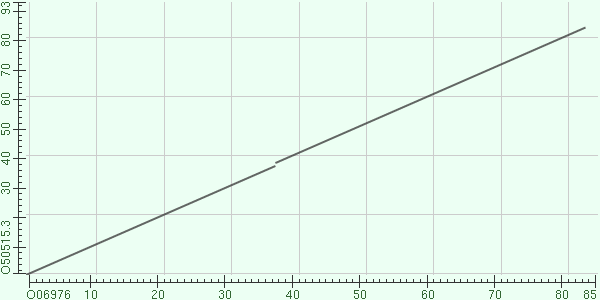
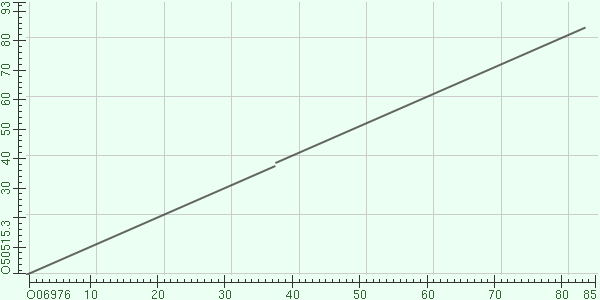
Кроме того, при помощи BLAST можно строить карты локального сходства двух последовательностей.
Все эти возможности описаны в соответствующих разделах практикума.
| Поиск по Swiss-Prot | Поиск по PDB | Поиск по "nr" | |
1. Лучшая находка (с последовательностью исходного белка) |
|||
| Accession | O06976.1 | 2RLZ_A | NP_391354.1 |
| E-value | 3e-54 | 3e-55 | 2e-52 |
| Вес (в битах) | 169 | 169 | 169 |
| Процент идентичности | 100% | 100% | 100% |
2. Число находок с E-value < 10–10 (хороших кандидатов в гомологи) |
36 | 33 | 1413 |
3. "Худшая из удовлетворительных" находка (последняя в выдаче с E-value < 1) |
|||
| Номер находки в списке описаний | 60 | 33 | 4160 |
| Accession | Q83QP3.1 | 1Y50_A | ZP_09377014.1 |
| E-value | 0.30 | 1e-12 | 0.98 |
| Вес (в битах) | 32.3 | 59.3 | 35.8 |
| % идентичности | 27 | 43 | 26 |
| % сходства | 47 | 67 | 47 |
| Длина выравнивания | 74 | 61 | 76 |
| Координаты выравнивания (от-до, в запросе и в находке) | В запросе: 10-81 В находке: 10-83 | В запросе: 1-61 В выравнивании:1-61 | В запросе: 10-83 В выравнивании: 10-85 |
| Число гэпов | 2 | 0 | 2 |
4. Число выводимых последовательностей-находок. |
|||
| Общее число находок | 66 (100 по умолчанию) | 37 (100 по умолчанию) | 4499 (разрешено 5000) |
| E-value последней находки | 8.3 (10 по умолчанию) | 9.2 (10 по умолчанию) | 9.9 (10 по умолчанию) |
| Поиск по Swiss-Prot (Actinobacteria) | Поиск по PDB (Сlostridia) | Поиск по "nr" (Eucarya) | |
Поиск ближайшего гомолога. |
|||
| Accession | O50515.3 | 3LE1_A | XP_003493001.1 |
| E-value | 1е-14 | 3e-29 | 6e-16 |
| Вес (в битах) | 58.2 | 98.2 | 74.3 |
| % идентичности | 38 | 58 | 42 |
| % сходства | 55 | 80 | 65 |
| Длина выравнивания | 84 | 77 | 83 |
| Координаты выравнивания (от-до, в запросе и в находке) | В запросе: 1-83 В находке: 1-84 | В запросе: 6-82 В выравнивании:6-82 | В запросе: 1-83 В выравнивании: 1-83 |
| Число гэпов | 1 | 0 | 0 |
| Рис 1.Карта локального сходства последовательностей с е-value=10 | Рис 2.Карта локального сходства при e-value=0.01 |
|
|