| H (BLOSUM62) | H (PHAT_T75_B73) | H (Sutormin) | ||
| Gly | G | -2 | -4 | -2 |
| Pro | P | -2 | -6 | -2 |
| Cys | C | -3 | -7 | -2 |
| Ser | S | -1 | -2 | -1 |
| Thr | T | -2 | -4 | -2 |
| Asn | N | 1 | 4 | 1 |
| Gln | Q | 0 | 2 | 1 |
| Asp | D | -1 | -1 | -1 |
| Glu | E | 0 | -1 | -1 |
| His | H | 8 | 11 | 8 |
| Arg | R | 0 | -4 | 0 |
| Lys | K | -1 | -5 | -1 |
| Ala | A | -2 | -3 | -2 |
| Met | M | -2 | -4 | -2 |
| Ile | I | -3 | -5 | -3 |
| Leu | L | -3 | -4 | -3 |
| Val | V | -3 | -5 | -3 |
| Phe | F | -1 | -2 | -1 |
| Trp | W | -2 | -3 | -1 |
| Tyr | Y | 2 | 3 | 1 |
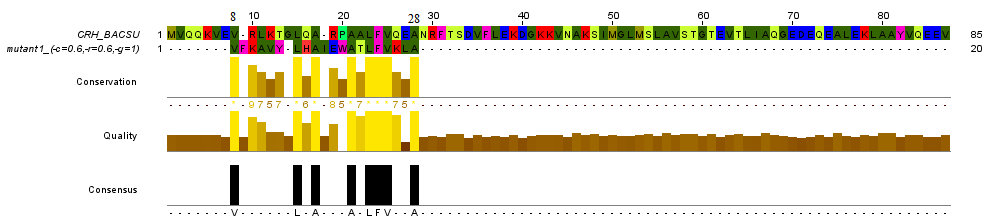
CRH_BACSU 1 MVQQKVEVRLKTGLQA-RPAALFVQEANRFTSDVFLEKDGKKVNAKSIMGLMSLAVSTGTEVTLIAQGEDEQEALEKLAAYVQEEV 85
|.....|.| ..|.|||:.|
mut1(0,6,0,6) 1 -------VFKAVYLHAIEWATLFVKLA----------------------------------------------------------- 20
CRH_BACSU 19 AALFVQEA 26
|.|||:.|
mut1(0,6,0,6) 13 ATLFVKLA 20
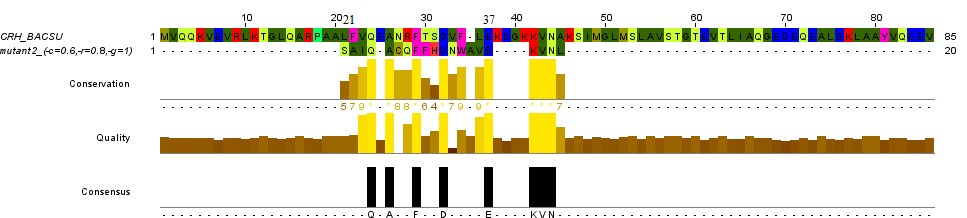
CRH_BACSU 1 ------------MVQQKVEVRLKTGLQARPAALFVQEANRFTSDVFLEKDGKKVNAKSIMGLMSLAVSTGTEVTLIAQGEDEQEALEKLAAYVQEEV 85
...:||.:
mut2(0,6,0,8) 1 SAIQACQFFHDNWAVEKVNL----------------------------------------------------------------------------- 20
CRH_BACSU 40 KKVN 43
:|||
mut2(0,6,0,8) 16 EKVN 19
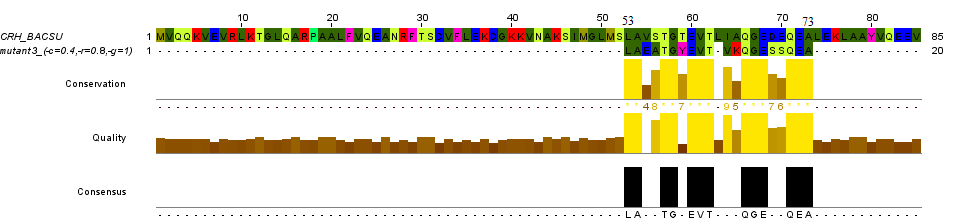
CRH_BACSU 1 MVQQKVEVRLKTGLQARPAALFVQEANRFTSDVFLEKDGKKVNAKSIMGLMSLAVSTGTEVTLIAQGEDEQEALEKLAAYVQEEV 85
||.:||.||| :.|||..|||
mut3(0,4,0,8) 0 ----------------------------------------------------LAEATGYEVT-VKQGESSQEA------------ 20
CRH_BACSU 53 LAVSTGTEVTLIAQGEDEQEA 73
||.:||.||| :.|||..|||
mut3(0,4,0,8) 1 LAEATGYEVT-VKQGESSQEA 20
| Общие проценты | % по участку выравнивания | |||||||
| Выравнивание вручную | needle | Выравнивание вручную | needle | |||||
| id | sim | id | sim | id | sim | id | sim | |
| mut1 | 9,2 | 11,5 | 9,3 | 10,5 | 38,1 | 47,6 | 38,1 | 42,9 | mut2 | 9,3 | 13,6 | 2,1 | 4,1 | 32,0 | 48,0 | 10 | 20 |
| mut3 | 15,3 | 16,5 | 15,3 | 17,6 | 61,9 | 66,7 | 61,9 | 71,4 |
Как видно из представленной таблицы, 1ое и 3е выравнивания (mut1 и mut3) были выровняны вручную примерно (или точно) так, как
это сделала программа needle. Отличия в similarity (sim) связаны с неоднозначнастью трактовки понятия "схожие по свойствам аминокислоты":
в нашем случае, я ориентировался на здравый смысл, а программа имела в виду матрицу BLOSUM62.
Результаты второго выравнивания сильно отличаются между ручным и машинным. Процент идентичности и схожести выше в случае ручного выравнивания,
однако вес машинного выравнивания выше (9 против 4). И все же, трудно предположить, чтобы скрипт сделал так много вставок (инсерций)
в самое начало последовательности и, скорее всего, это как-раз тот случай, когда правильное выравнивание является
с точки зрения программы не самым оптимальным. Возможно, если задавать другие параметры для гэпов, то "правильное" выравнивание
все-таки будет найдено.
Правильность ручных выравниваний подтверждаются программой water, которая определила участки выравнивания,
совпадающие или перекрывающиеся с найденными вручную. Так для второго выравнивания,
найднный фрагмент является частью не машинного, а "ручного" выравнивания, что делает ручное выравнивание более предпочтительным.