




При помощи пакета 3DNA можно построить 46 моделей ДНК с различными параметрами. В нашей работе были построены
A-, B- и Z-формы ДНК. Команда fiber позволяет не только указать тип модели, но и во многих случаях желаемую последовательность.
Исключение составляет Z-форма ДНК, моделирование которой возможно только для пар GC.
Результатом работы программы является pdb файлы моделей. Файлы были открыты и обработаны в Jmol, результаты работы
представлены на рисунках 1-6.
|
|
|
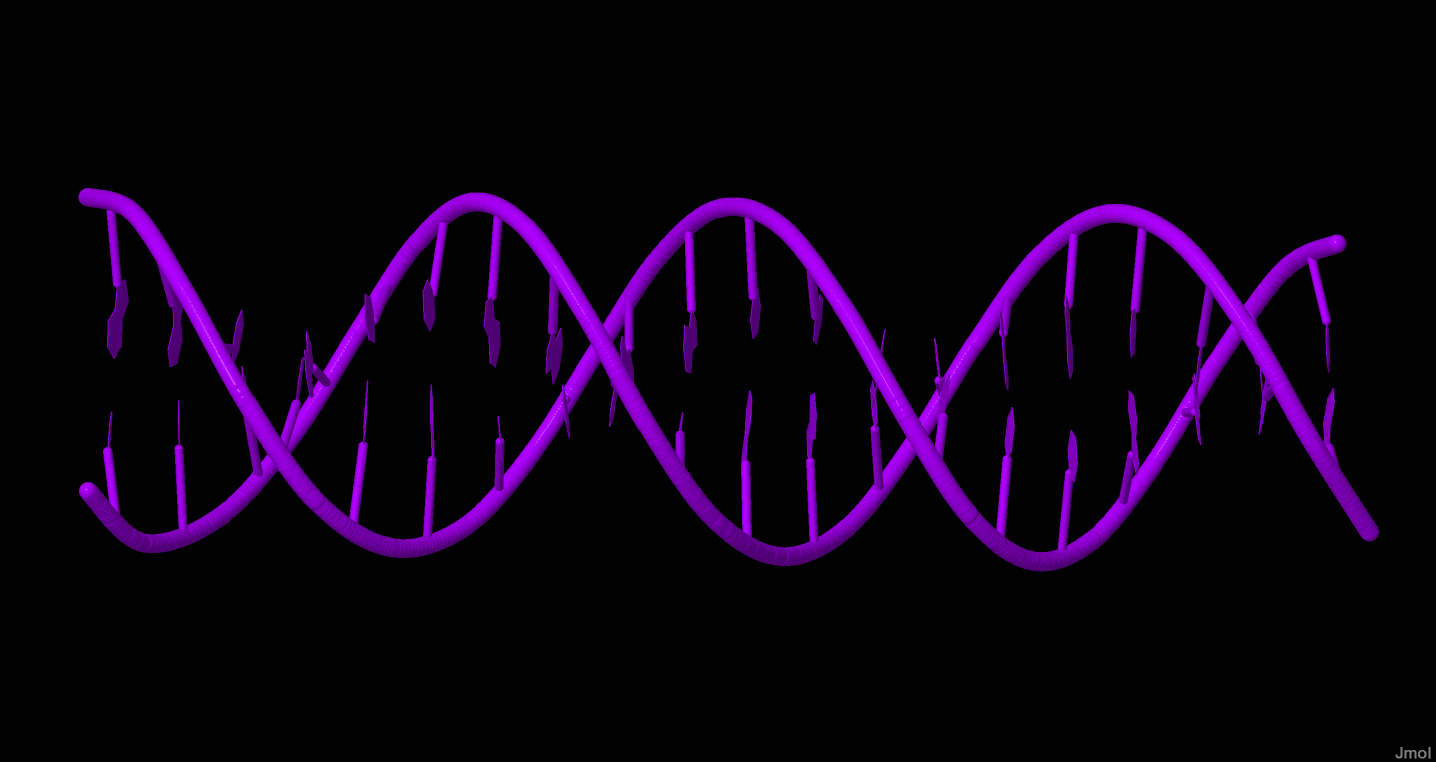
| Рис 1. А-форма ДНК вид сбоку. | Рис 2. В-форма ДНК вид сбоку. | Рис 3. Z-форма ДНК вид сбоку. |
|
|
|
| Рис 4. А-форма ДНК вид с торца. | Рис 5. В-форма ДНК вид с торца. | Рис 6. Z-форма ДНК вид с торца. |
На представленных рисунках видны основные отличия форм ДНК: А-форма имеет большую полость в центре, спираль правая, плоскости оснований не параллельны друг-другу. B-форма также правозакрученная, но нет полости, основания параллельны. Z-форма левая, полость есть, но маленькая, цепи выглядят как-будто изломанными.
На примере модели A-формы ДНК, покажем возможности Jmol для визуализации пространственных структур ДНК. В качестве задачи, попробуем тем или иным способом выделить следующие элементы структуры:
В результате, были получены следующие изображения:
 |
|
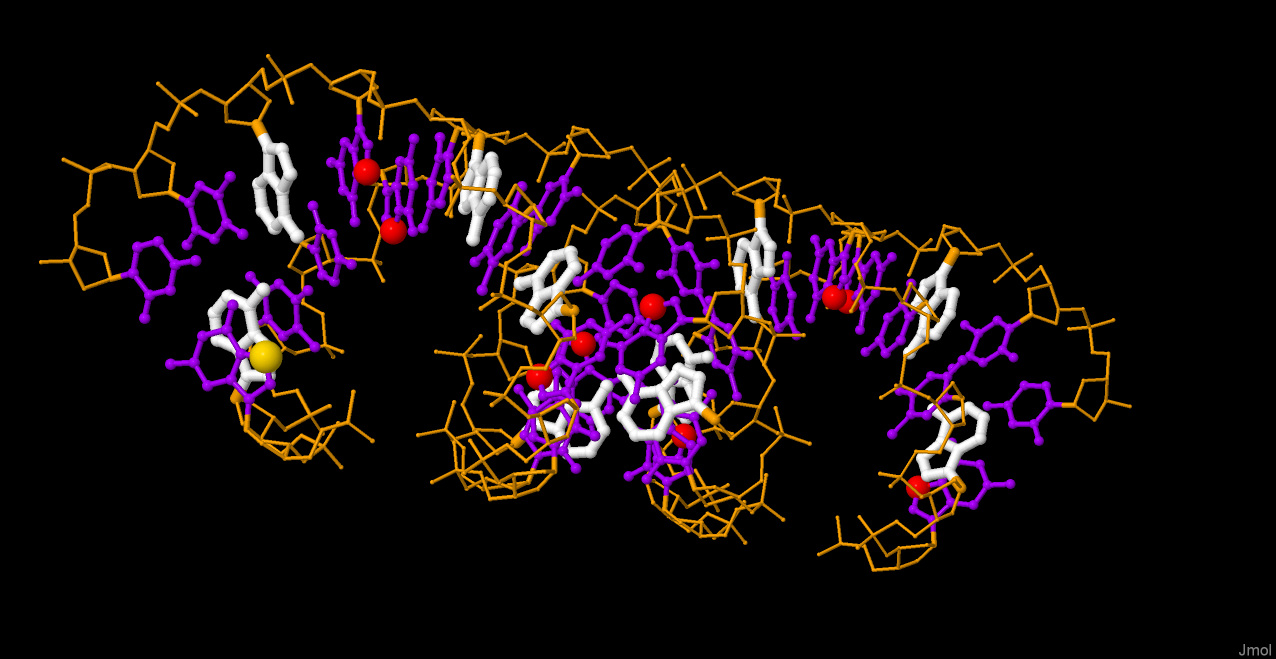
| Рис 1. Модель A-формы ДНК, обработанная в Jmol. | 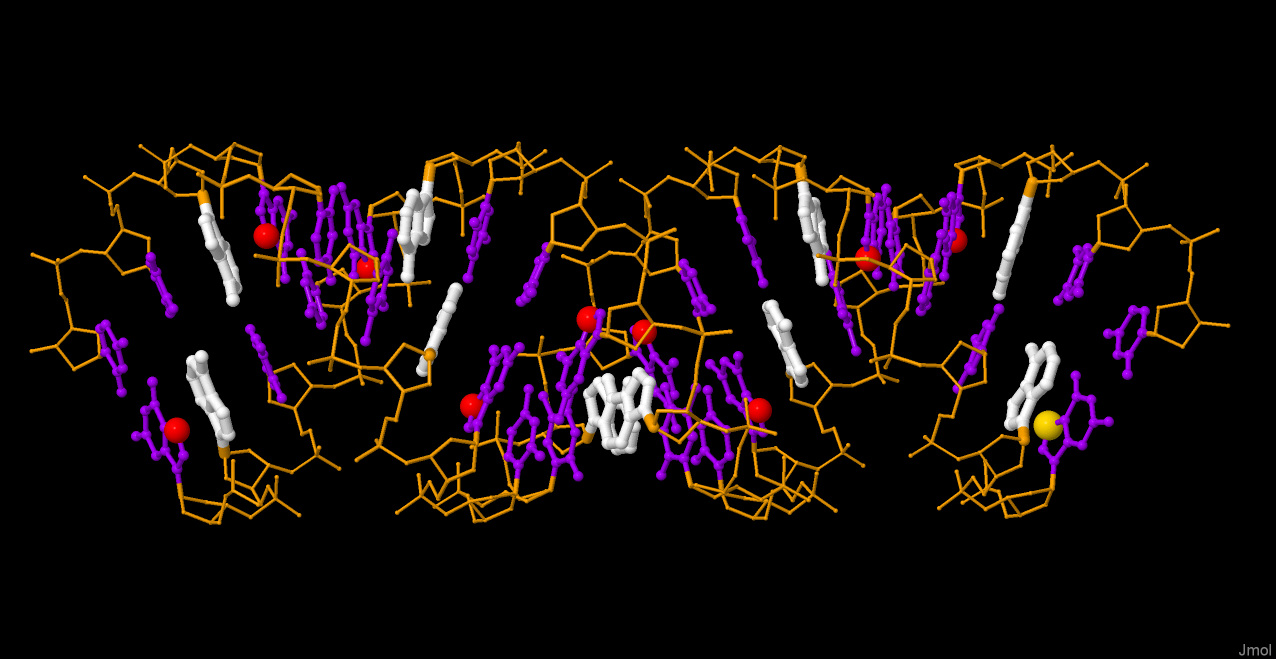 |
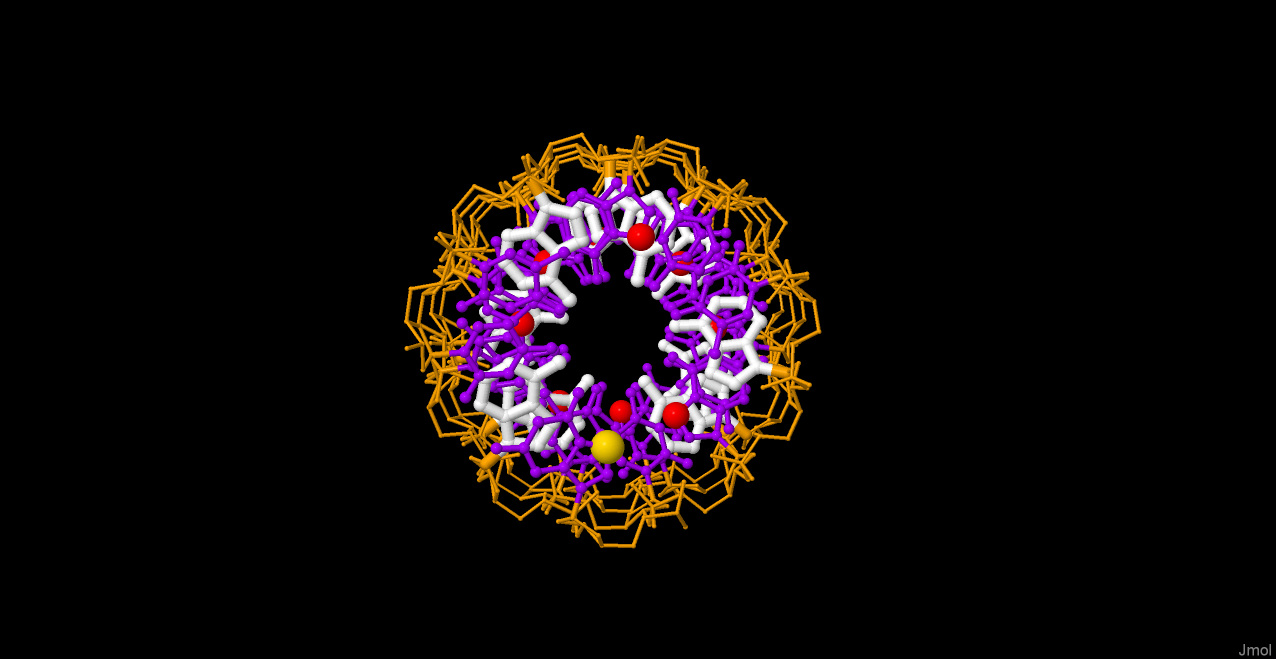 |
| Рис1,2,3. Остов ДНК выделен оранжевым, связи и атомы тонкие и маленькие. Атомы оснований имеют больший радиус, все, кроме аденинов фиолетовые. Аденины выделены белым, связи по толщине как атомы. Седьмые атомы гуанинов красные и имеют большой радиус. Седьмой атом преого гуанина еще большего радиуса и имеет желтый цвет. |
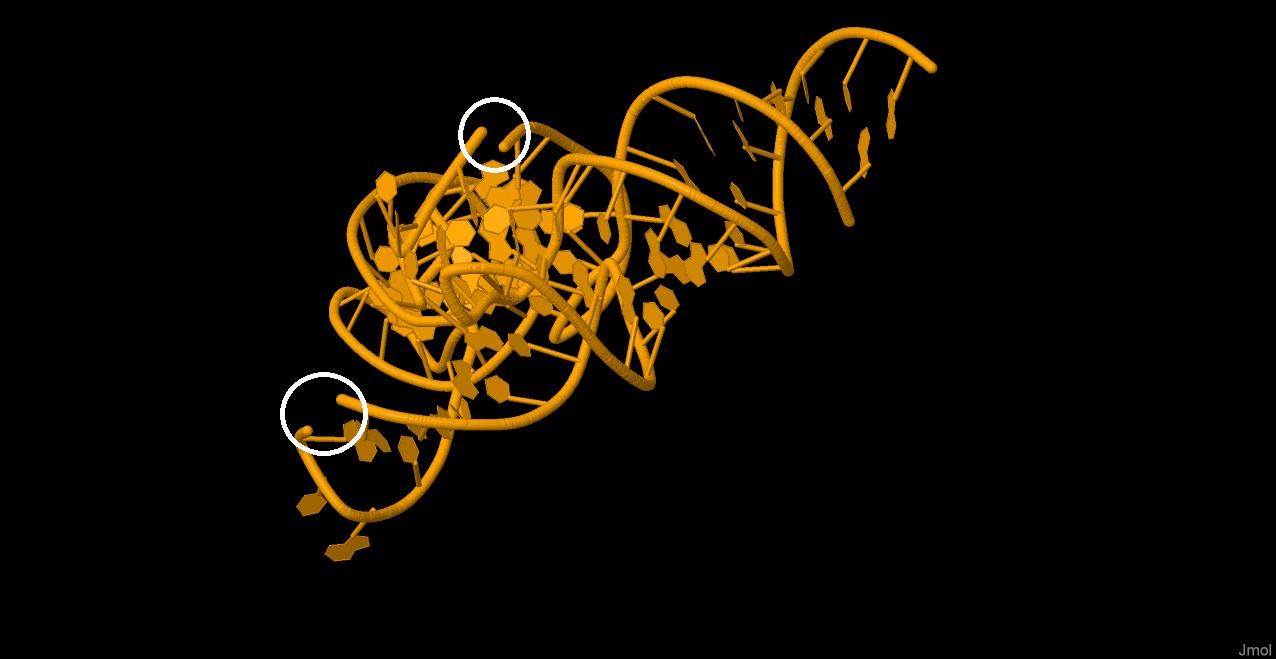
Были рассмотрены следующие структуры: 1h3e (тРНК) и 1pp7 (ДНК-белковый комплекс). Для них, средствами Jmol были получены изображения только нуклеиновых кислот.
 |
 |
| Рис 1,2. тРНК. Визуализация cartoon. Разрывы в цепях обведены белыми кружками. |  |
Рис 3. ДНК. Визуализация cartoon. В цепях нет видимых разрывов. |
В структуре тРНК были обнаружены два разрыва. Однако при внимательном рассмотрениии и при визуализации всех атомов и связей тРНК, было обнаружено, что разрывов нет. Дело в том, что в данных сайтах располагаются модифицированные основания - C и U, не имеющие нуклеотидов.

Рис 4. Структура РНК. Места "разрывов" имеют серый цвет.
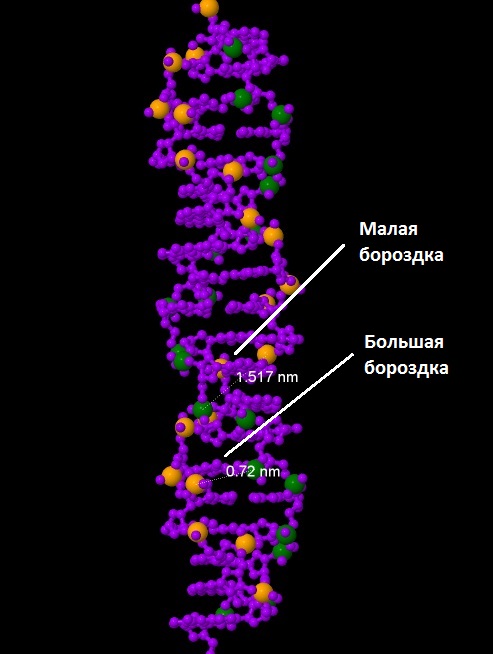
Как известно, двойная спираль ДНК имеет в своей структуре большую и малую бороздки (см. рис. 1). В В-форме ДНК для тимина были определены атомы, экспонированные в ту или иную бороздки (см. рис. 2).
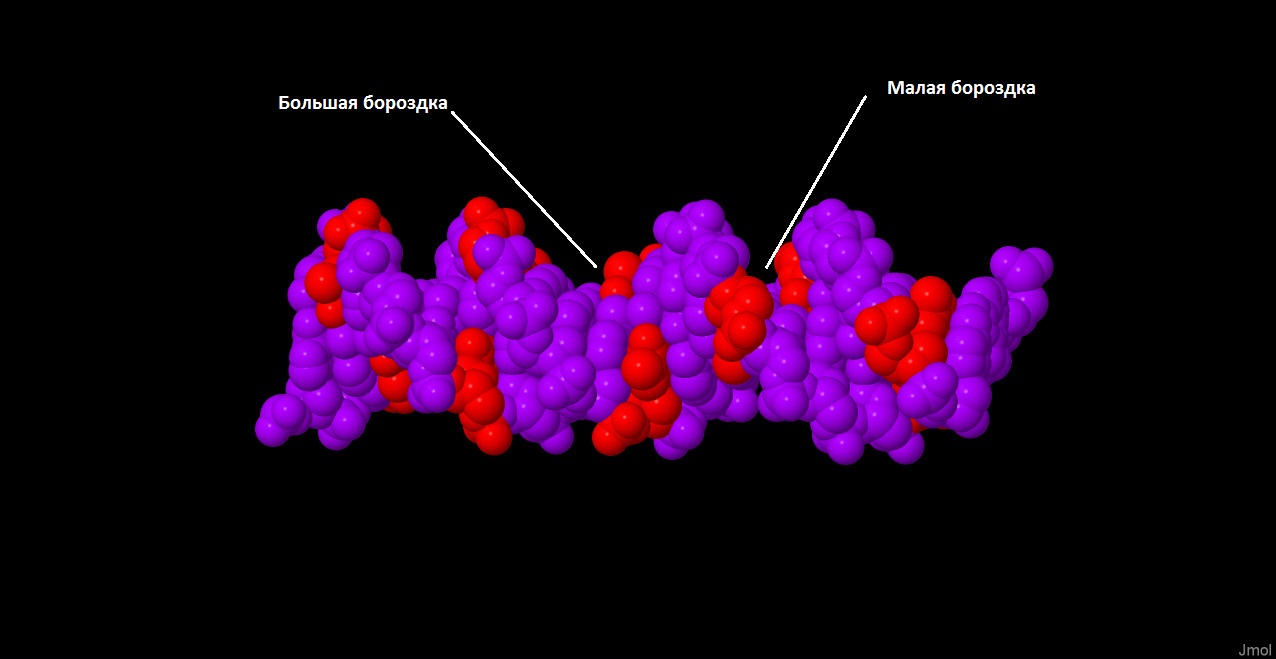
Рис 1. На данном рисунке, на модели ДНК отмечены большая и малая бороздки.
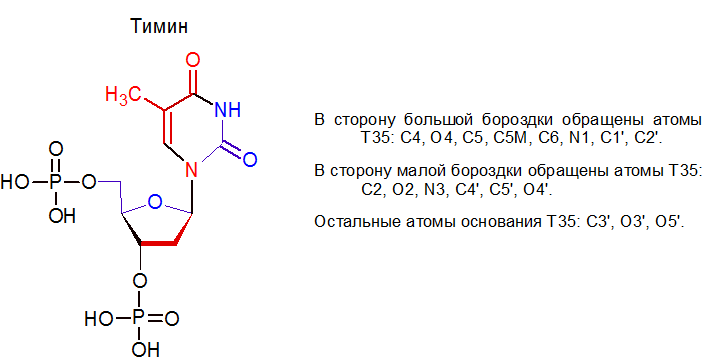
Рис 2. Красным цветом выделены атомы, экспонированные в большую бороздку, синим - в малую.
Ниже представлены основные параметры разных форм ДНК с соответствующими иллюстрациями.
| В-форма | A-форма | *Z-форма | |
| Тип спирали (правая или левая) | Правая | Правая | Левая |
| Шаг спирали (nm) | 3,091 (по фосфорам) | 2,803 (по фосфорам) | 4,35 (по фосфорам) |
| Число оснований на виток | 10 | 12 | 13 |
| Ширина большой бороздки (nm) | 1,721 | 0,798 | 0,72 |
| Ширина малой бороздки (nm) | 1,169 | 1,681 | 1,517 |
| B-форма | 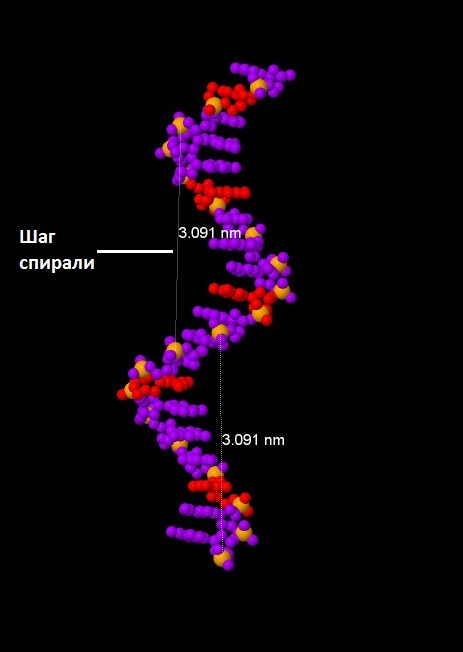 |
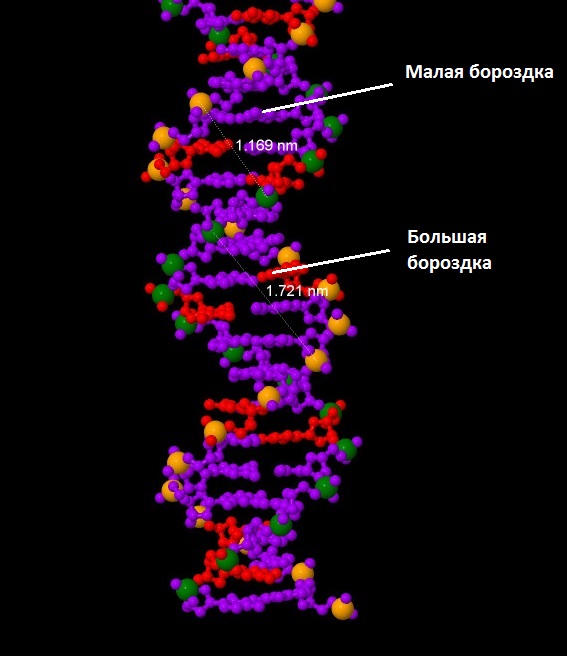 |
|
| A-форма | 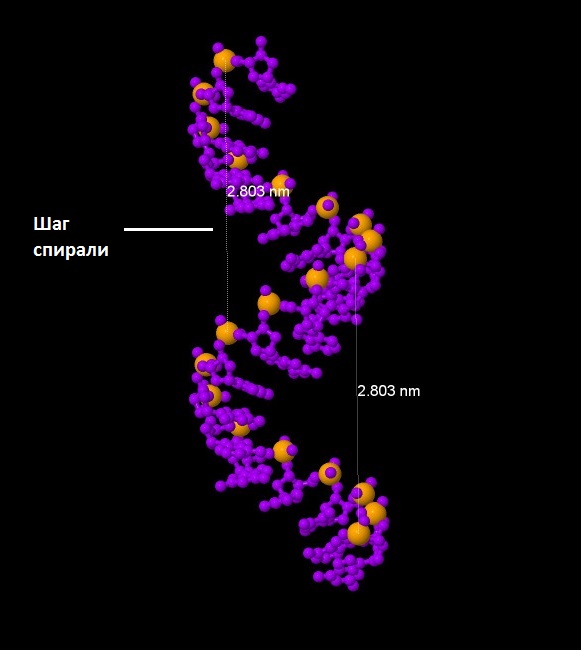 |
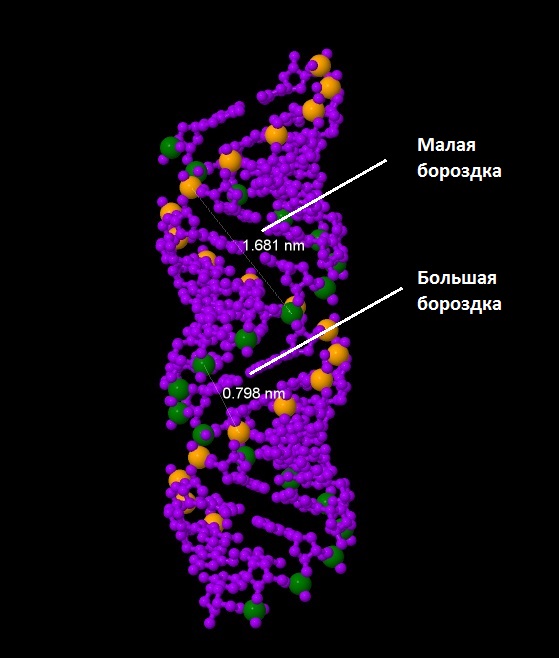 |
|
| Z-форма |  |
 |
Исследование показало, что самая широкая бороздка не обязательно является большой (для A и Z-форм). Дело в том, что по определению, большая бороздка, это - самая глубокая бороздка.
Торсионный угол, т.е. угол поворота связи А-В вокруг связи В-С относительно связи С-D,
определяется как угол между плоскостями, содержащими атомы А, В, С и атомы B, C, D.
В случае нуклеотидов можно определить множество торсионных углов между связями атомов в рибозе и фосфате.
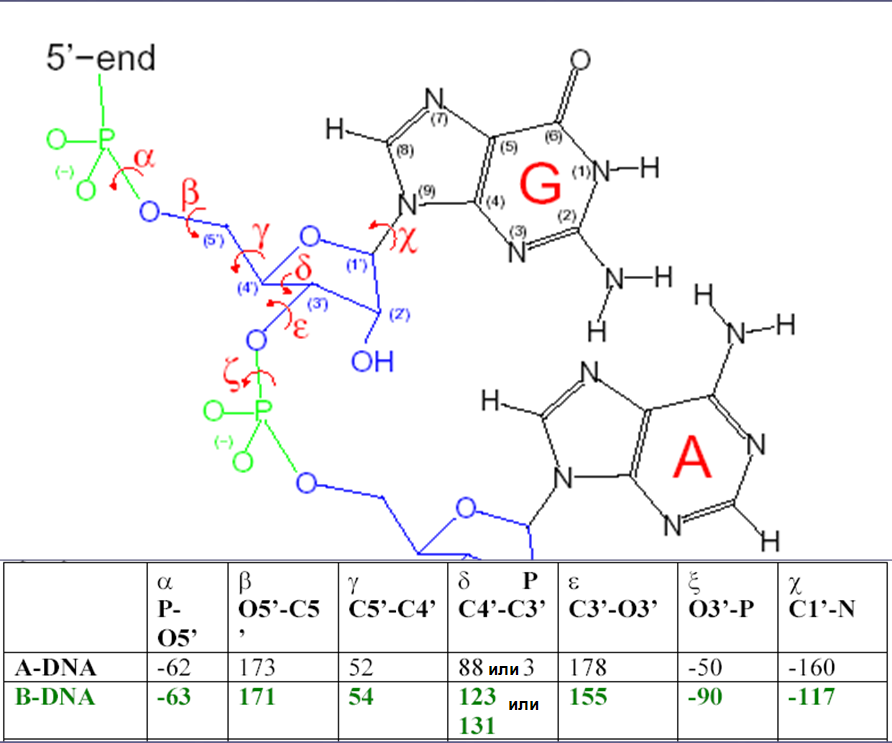
Рис. 1. Расположение, названия и значения торсионных углов на примере гуанина.
Средства Jmol позволяют определять торсинные углы заданной последовательности из четырех последовательно соединенных атомов.
В нашей работе определялись торсионные углы тимина. Результаты работы представлены в таблице:
| α O3'(n+1)-P(n)-O5'(n)-C5'(n) | β P(n)-O5'(n)-C5'(n)-C4'(n) | γ O5'(n)-C5'(n)-C4'(n)-C3'(n) | δ C5'(n)-C4'(n)-C3'(n)-O3'(n) | ε C4'(n)-C3'(n)-O3'(n)-P(n-1) | ξ C3'(n)-O3'(n)-P(n-1)-O5'(n-1) | χ C2(n)-N1(n)-C1'(n)-O4'(n) | |
| А-форма ДНК | -51.7 ° | 174.8 ° | 41.7 ° | 79.1 ° | -147.8 ° | -75.1 ° | -157.2 ° |
| В-форма ДНК | -29.9 ° | 136.3 ° | 31.2 ° | 143.3 ° | -140.8 ° | -160.5 ° | -98.0 ° |
Также были получены изображения тимина с двумя соседними нуклеотидами. Без соседних нуклеотидов невозможно определение совместных углов: α, ε, ξ
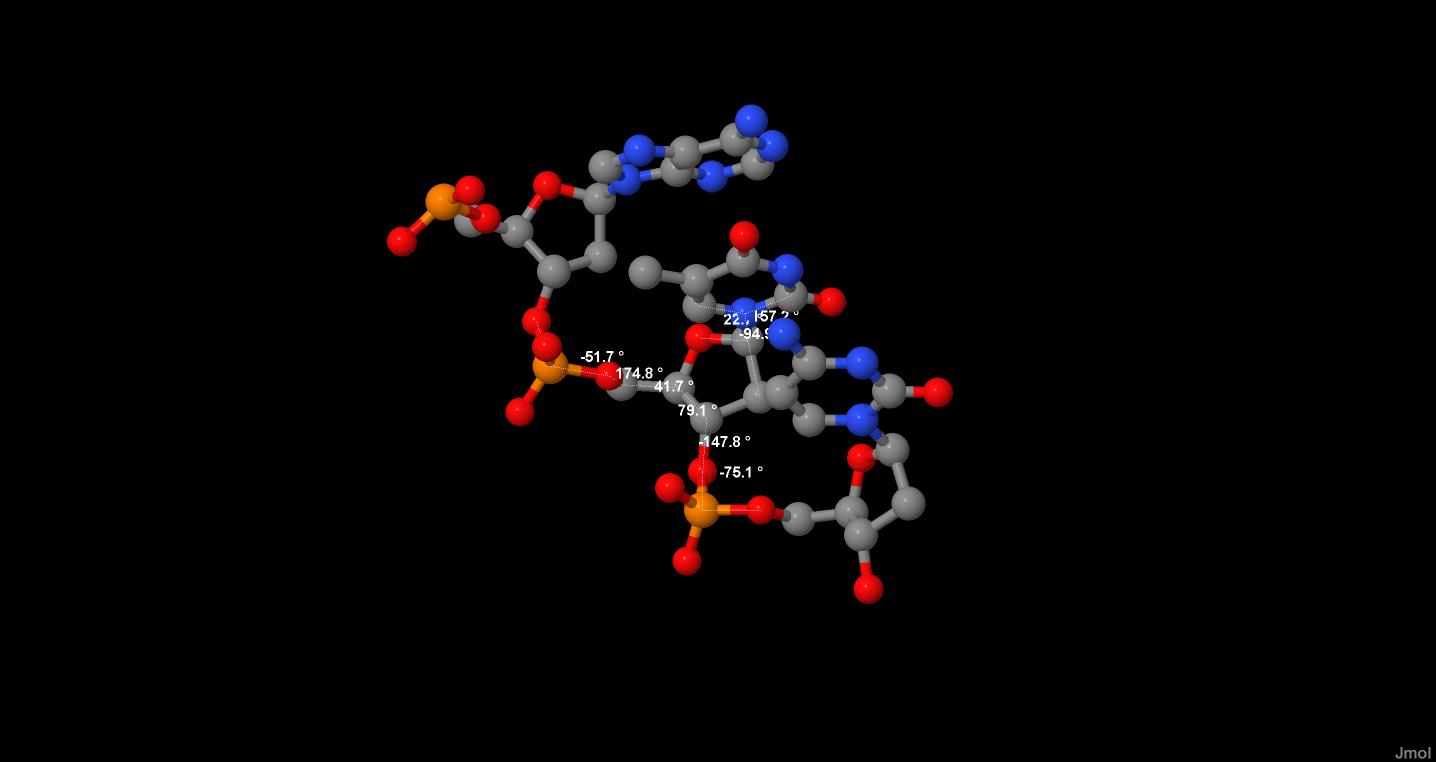
Рис. 2. Расположение и значения торсионных углов для тимина в А-форме ДНК.
На рисунке 2 заметно, что в области угла χ подписано несколько значений. Это связано с неоднозначностью определения данного угла. Дело
в том, что можно по разному выбрать последовательность из четырех атомов: C6-N1-C1'-C2' или C6-N1-C1'O4'
или C2-N1-C1'-C2' или C2-N1-C1'-O4'.
Для унификации предлагается пользоваться старшинством атомов и групп: рассматривать старшинство по кругам, по массе атомов.
В соответствии с этим правилом следует выбрать последовательность C2-N1-C1'-O4' т.к.
С2 соединен с кислородом - он тяжелее, а O4' сам по себе тяжелееС2'.
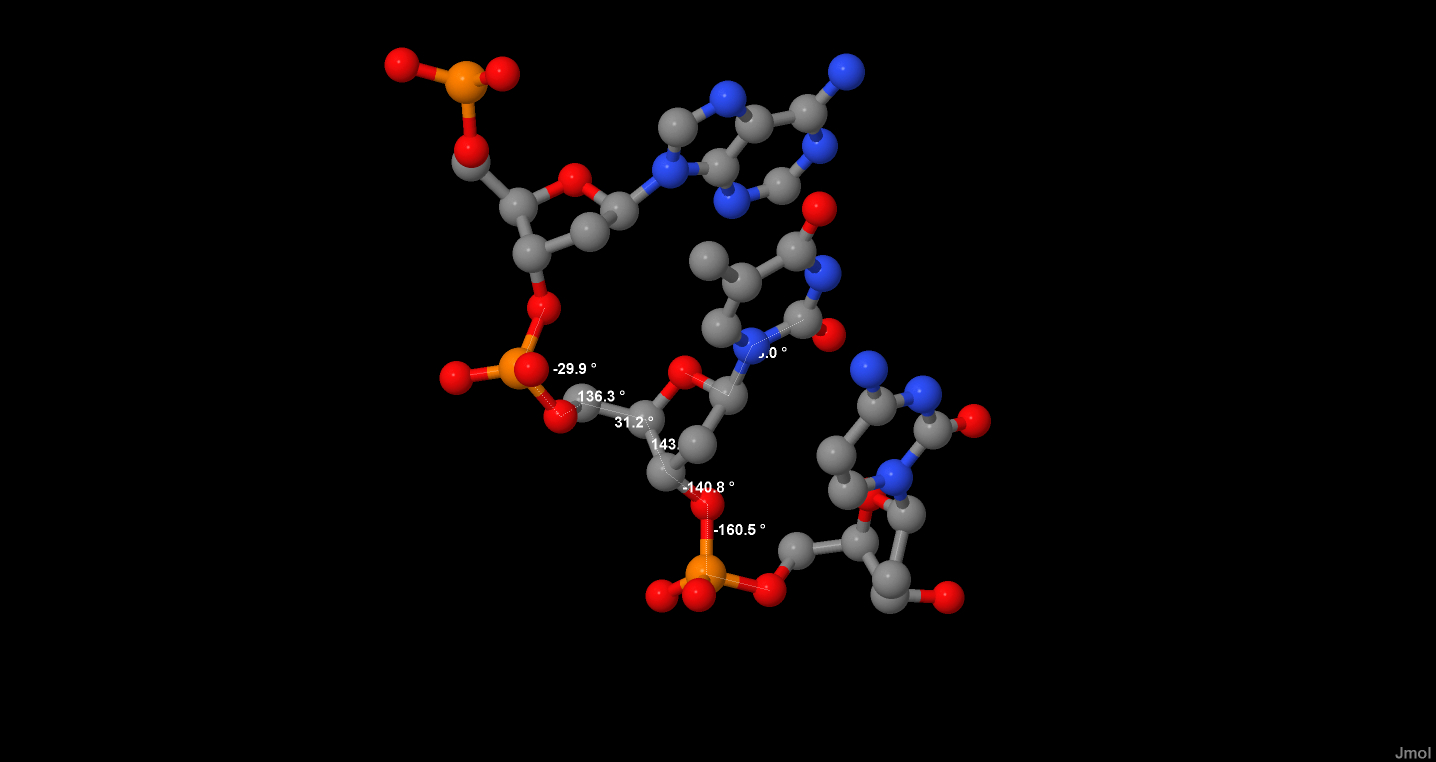
Рис. 2. Расположение и значения торсионных углов для тимина в B-форме ДНК.
Торсионные углы можно рассчитать при помощи программ пакета 3DNA. Для примера, мы получим торсионные углы для РНК (из 1h3e) и ДНК (из 1pp7).
Однако, для этого необходимо сначала подготовить файлы.
В первую очередь, надо получить pdb-файл только для нуклеиновой кислоты (без белка). Это можно сделать простым копированием
той части документа, которая содержит координаты и параметры интересующих нас атомов.
Вторым пунктом, надо перевести файл в "старый" формат pdb, т.к. программы пакета 3DNA понимают только его.
Это можно сделать программой remidiator:
remediator --old ''XXXX.pdb'' > ''XXXX_old.pdb
Следующий шаг: подготовить полученый "old" файл для программы analyze, которая и считает торсионные углы. Это делается программой
find_pair. Так как эти программы из одного пакета и совместимы друг с другом, то удобно объединят их в конвеер:
find_pair -s XXXX_old.pdb stdout | analyze
На выходе мы получаем множество файлов с различными данными. Нас интересует файл - XXXX_old.outs. Помимо всего прочего,
там можно найти таблицу с торсионными углами. Причем, там есть 2 разные таблицы - одна для остова и угла хи, а другая - для сахара.
В таблице 1 представлены торсионные углы для ДНК из структуры 1pp7.

В конце-концов при помощи Excel были просчитаны средние значения для углов:
| α | β | γ | δ | ε1 | ξ | χ |
| -54,3 | 36,4 | 44,9 | 137,3 | -162,1 | -81,5 | -106,9 |
Таблица 1. Средние значения торсионных углов для ДНК из 1pp7
Таблица 3. Средние значения торсионных углов для РНК из 1h3e.
Отклонение угла от среднего определялось как разность модулей этих углов. Интересно, что максимально деформированные углы
группируются вместе, а не разбросаны равномерно по всей последовательности. Вероятно такие участки являются наиболее напряженными
элементами структуры РНК.
Пространственная организация РНК гораздо сложенее, чем у ДНК. У РНК можно выделить: стебли, петли, выпетливания, псевдоузлы и т.д.
На примере нашей РНК попробуем определить стебли, т.к. это проще всего сделать алгоритмически: необходим некий участок - дуплекс, имеющий
примероно спиральную структуру, в котором цепи антипараллельны. l
Очевидно, среди этих участков и нужно искать стебли.
Уже знакомая нам программа find_pair позволяет находить и неканонические пары оснований. Эта информация представлена в файлах, описывающих
спиральные элементы в структуре.
Очевидно, что в пары оснований в стеблях соединены водородными связями. Однако, весь ассортимент водородных связей на этом не заканчивается,
они есть и вне этих элементов, дополнительно стабилизируя пространственную структуру.
Рис 1. Все пары оснований в структуре РНК из 1h3e. Охристым цветом выделены пары оснований, образующие стебли.
Для нуклеиновых кислот характерен особый тип внутри- и межмолекулярного взаимодействия. Оно получило название stacking-взаимодействия и основано
на перекрывании пи-систем ароматических структур - азотистых оснований. Как известно, такие структуры плоские, а орбитали пи-системы
находятся над и под циклами. Соответственно, если 2 а.о. лежат друг над другом и при этом достаточно близко, то происходит взаимодействие
пи-систем, дополнительно стабилизирующее структуру. Рис. 2.Таблица из файла XXXX_old.out с информацией о перекрывании пар оснований. Голубым выделены строчки пар с наибольшим
перекрыванием.Охристым цветом - с нулевым перекрыванием, зеленым - со средним перекрываниемю
Аналогичные операции были проделаны с РНК.
Полная таблица 2 торсионных углов для РНК из 1h3e.
α
β
γ
δ
ε1
ξ
χ
-35,1
16,3
63,6
91,8
-144,0
-56,4
-132,5
Для данной структуры можно попробовать определить самый деформированный нуклеотид. Это можно сделать, сравнив его торсионные углы с их
средними значениями (таблица 4). На взгляд, это 48 U.
α
β
γ
δ
ε1
ξ
χ
РНК среднее
-35,1
16,3
63,6
91,8
-144,0
-56,4
-132,5
U48
151,3
-167
175,1
88
-179,5
111,3
-87,7
Таблица торсионных углов с просчитаными отклонениями от среднего. Красным цветом выделены ячейки, значения в которых
отличны по знаку от среднего значения. Зеленым цветом - наибольшие отклонения от среднего.
Определение элементов вторичной структуры РНК
Определение стеблей производится при помощи все той же программы find_pair, которая находит спиральные регионы в структуре:
find_pair -d XXXX_old.pdb XXXX_old.inp
Опять генерируется много-много разных файлов с разнообразной информацией о структуре. Нам нужны файлы: bp_order.dat и файлы типа -
XXXX_old.inp_000n, где n - числа начиная с 1.
В bp_order.dat сплошным списком идут пары оснований, образующие спиральные структуры.
В файлах типа XXXX_old.inp_000n - отдельные спиральные структуры.
Судя по выводу программы, в данной РНК таких участков:
(Совершенно не понятно, откуда взялись номера - 75-78, если в последовательности РНК всего 74 основания?)
Дело в очень странной нумерации нуклеотидов: после номера 47 идет 47A, 47B, 47C, 47D, 47E, 47F, 47G, 47H, 47I и только потом - 48!
Дальнейший отбор проводился визуально в Jmol. Результаты:
Неканонические пары оснований в РНК
Всего программой было обнаружено 9 не Уотсон-Криковских пар:
4 неканонически связанных UA, 3 UG, 1 CG и 1 СА.
Водородные связи в РНК
Как можно найти такие связи? Первый вариант - проанализировать структуру в Jmol. Однако есть и второй, програмный.
Используя программу find_pair можно найти все представленные в структуре пары оснований:
find_pair -z rna_old.pdb stdout | analyze
Следовательно, если вычесть из множества всех пар, те, которые в стеблях, мы получим искомый набор.
Список всех пар нуклеотидов можно найти в файле XXXX_old.out. и на рисунке 1. 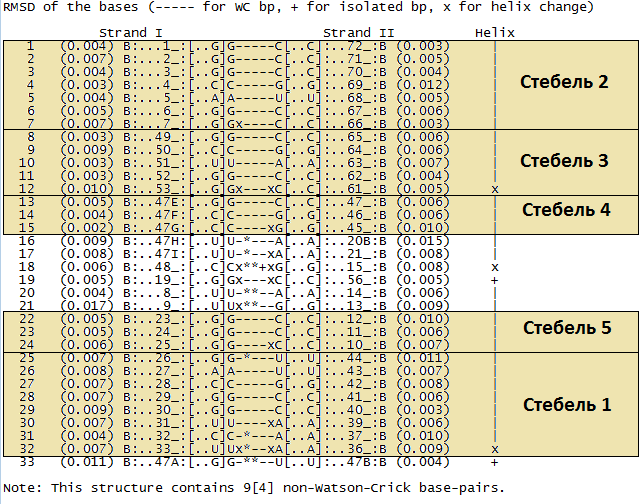
В соответствии с идеей, остальные пары оснований (не залитые охрой) не входят в стебли, и дополнительно стабилизируют структуру молекулы.
Стэкинг-взаимодействия в РНК
Информацию о стэкинге можно получить все той же программой find_pair:
find_pair -z rna_old.pdb stdout | analyze
В выходном файле XXXX_old.out можно найти информацию о последовательном перекрывании пар оснований (Рис 2.). Логично предположить, что чем
больше пары перекрываются, тем сильнее stacking. 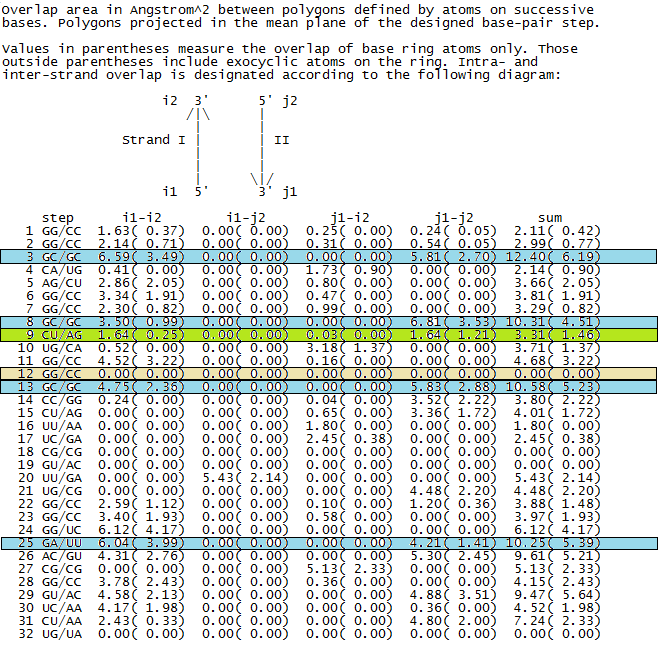
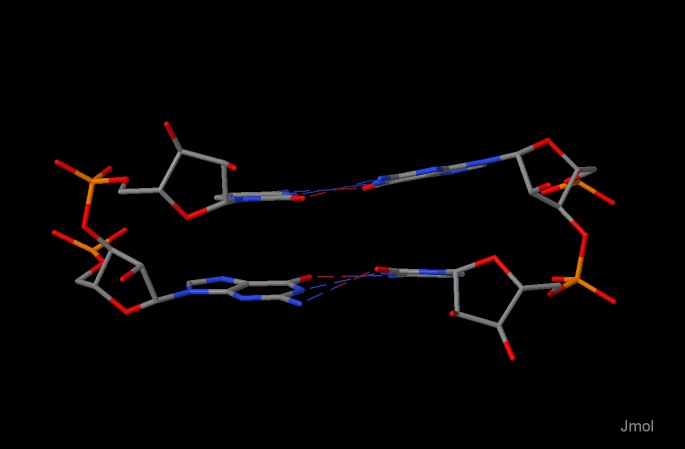
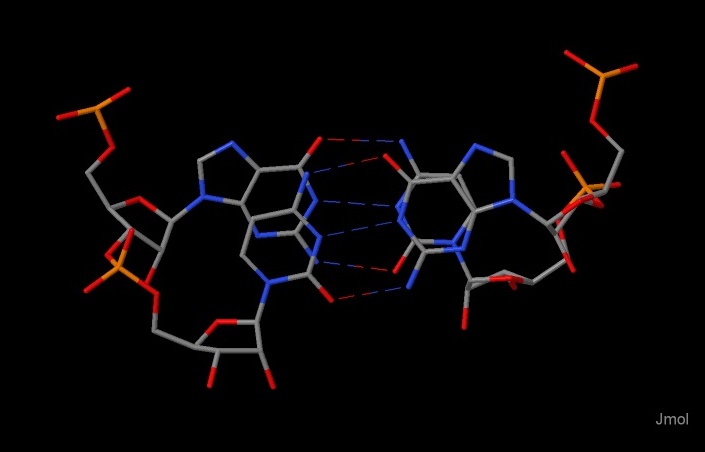
В дальнейшем, мы будем работать с 3ей парой, имеющей максимальное перекрываение - 12,4 квадратных ангстрем.
Информацию о пространственной структуре интересующей нас пары можно получить из файла stacking.pdb который генерируется одновременно с остальными
файлами программой find_pair. В этом файле есть pdb модели всех перекрывающихся попарно пар.
Для того, чтобы получить только 1 интересную нам структуру можно просто копировать нужный блок в новый файл.
Открыв этот файл в Jmol можно внимательно рассмотреть полученую область РНК со стэкингом (Рис 2., Рис 3.)
Рис 2,3. Визуализация перекрывающихся пар в Jmol. Водородные связи между основаниями показаны пунктиром.
Аналогичную работу можно проделать при помощи программ:
ex_str -N stacking.pdb stepN.pdb
Где N - номер блока с нужной парой.
Полученый pdb-файл можно конвертировать в формат ps:
stack2img -cdolt stepN.pdb stepN.ps
Однако с полученым файлом stepN.ps возникают проблемы - непонятно, как его открывать. Сделать это можно, на пример, в Photoshop, где его можно
конвертировать в jpg (Рис 4.).
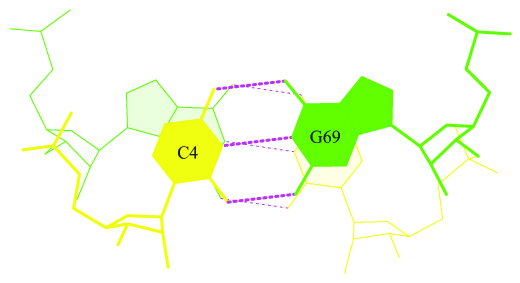
Рис 4. Перекрывание пар оснований. Визуализация stack2img.
Аналогично было получено изображение пары с наименьшим перекрыванием (рис 5.)
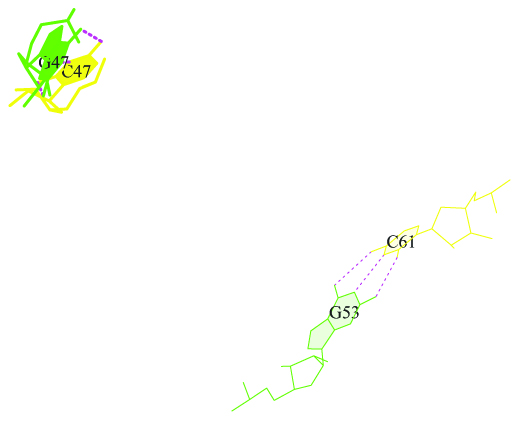
Рис. 5. Пара с нулевым перекрыванием.
Как видно из рисунка 5, пары просто разнесены слишком далеко друг от друга.
Для контроля были выбраны пары с небольшим перекрыванием (рис 6.)
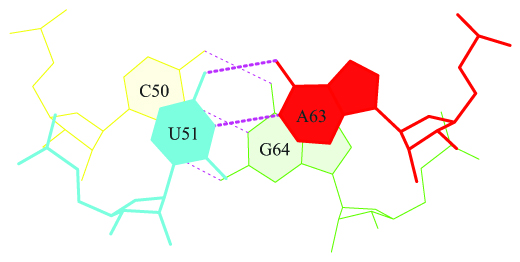
Рис. 6. Пара с небольшим перекрыванием.