- Краткое описание структуры в файле 1QRT.pdb
В файле приведены координаты атомов двух молекул:
- молекулы белка - синтетазы глютаминовой тРНК (GLUTAMINYL-TRNA SYNTHETASE, или GLUTAMINYL-TRNA LIGASE) из ESCHERICHIA COLI;
- молекула глютаминовой тРНК (TRNAGLN2, или GLUTAMINE TRANSFER RNA) из ESCHERICHIA COLI.
Для исследования была выбрана цепь B, представляющая собой глютаминовую тРНК со следующей последовательностью:
[2] 5’ –GGGGUAUCGCCAAGCGGUAAGGCACCGGAUUCUGAUUCCGGCAUUCCGAGGUUCGAAUCCUCGUACCCCAGCCA – 3’[76], где 2 и 76 - номера первого и последнего нуклеотида.
В последовательности на 3'-конце имеется триплет CCA, к которому собственно и присоединяется аминокислота. Координаты его атомов можно найти в соответствующем pdb-файле.
- Исследование вторичной структуры
С помощью программы find_pair пакета 3DNA были определены возможные водородные связи между азотистыми основаниями (1QRT.fp).
В соответствии с полученными данными:
- акцепторный стебель состоит из участка 2-7 и комплементарного ему участка 66-71,
- Т-стебель – из участков 49-53 и 61-65,
- D-стебель - из участков 10-12 и 23-25
- антикодоновый бель – из участков 37-44 и 26-33.
Ниже представлен скрипт для получения в RasMol изображения остова исследуемой тРНК, где акцепторный стебель выделен красным, Т-стебель - зеленым, D-стебель - синим, антикодоновый - оранжевым.
| Рис.1. Вторичная структура молекулы аптамера глютаминовой тРНК из ESCHERICHIA COLI | Скрипт для получения изображения |
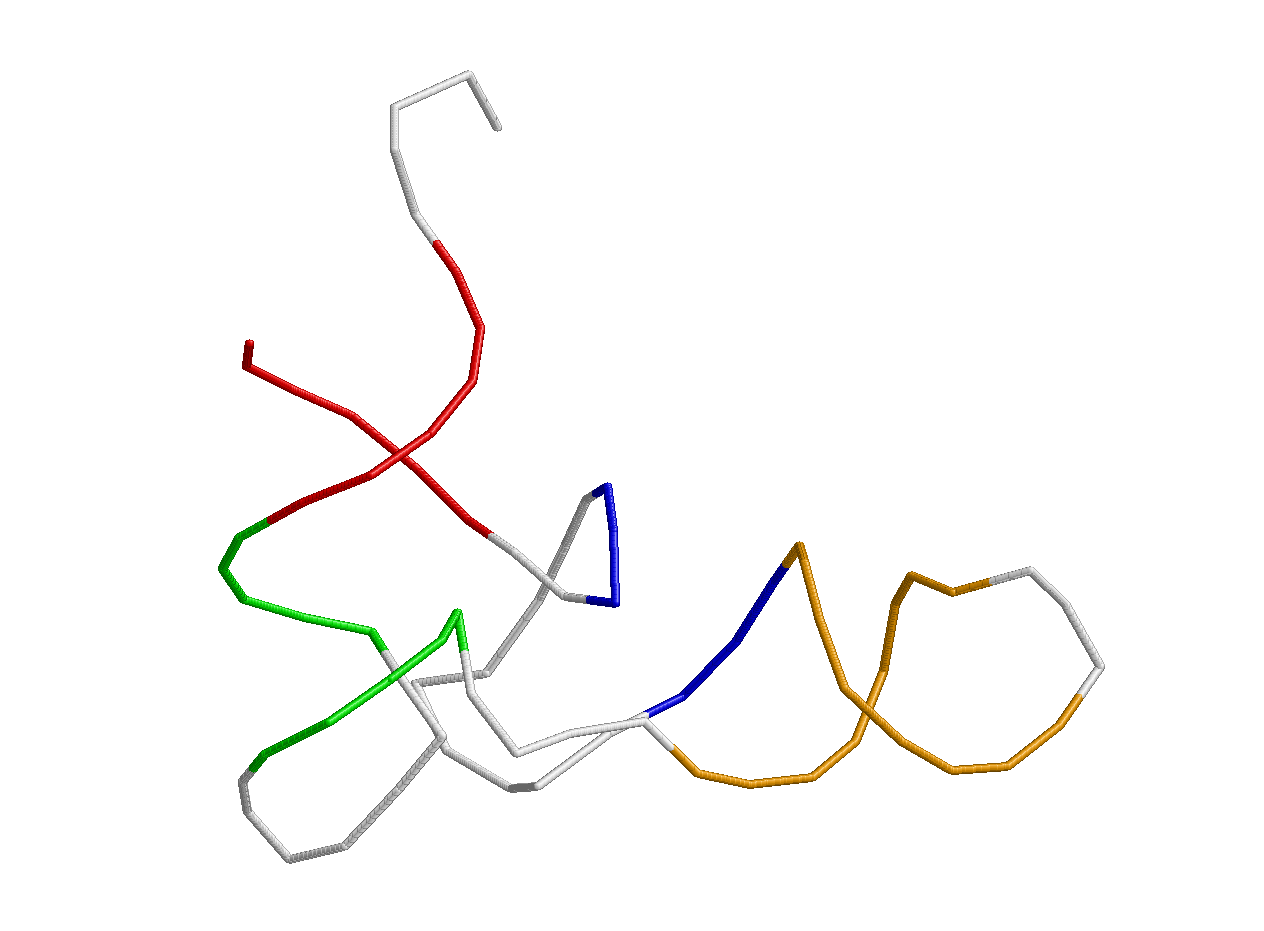 |
background white restrict none select *:B backbone 75 color white select 2-7 or 66-71 color red select 49-53 or 61-65 color green select 10-12 or 23-25 color blue select 37-44 or 26-33 color orange |
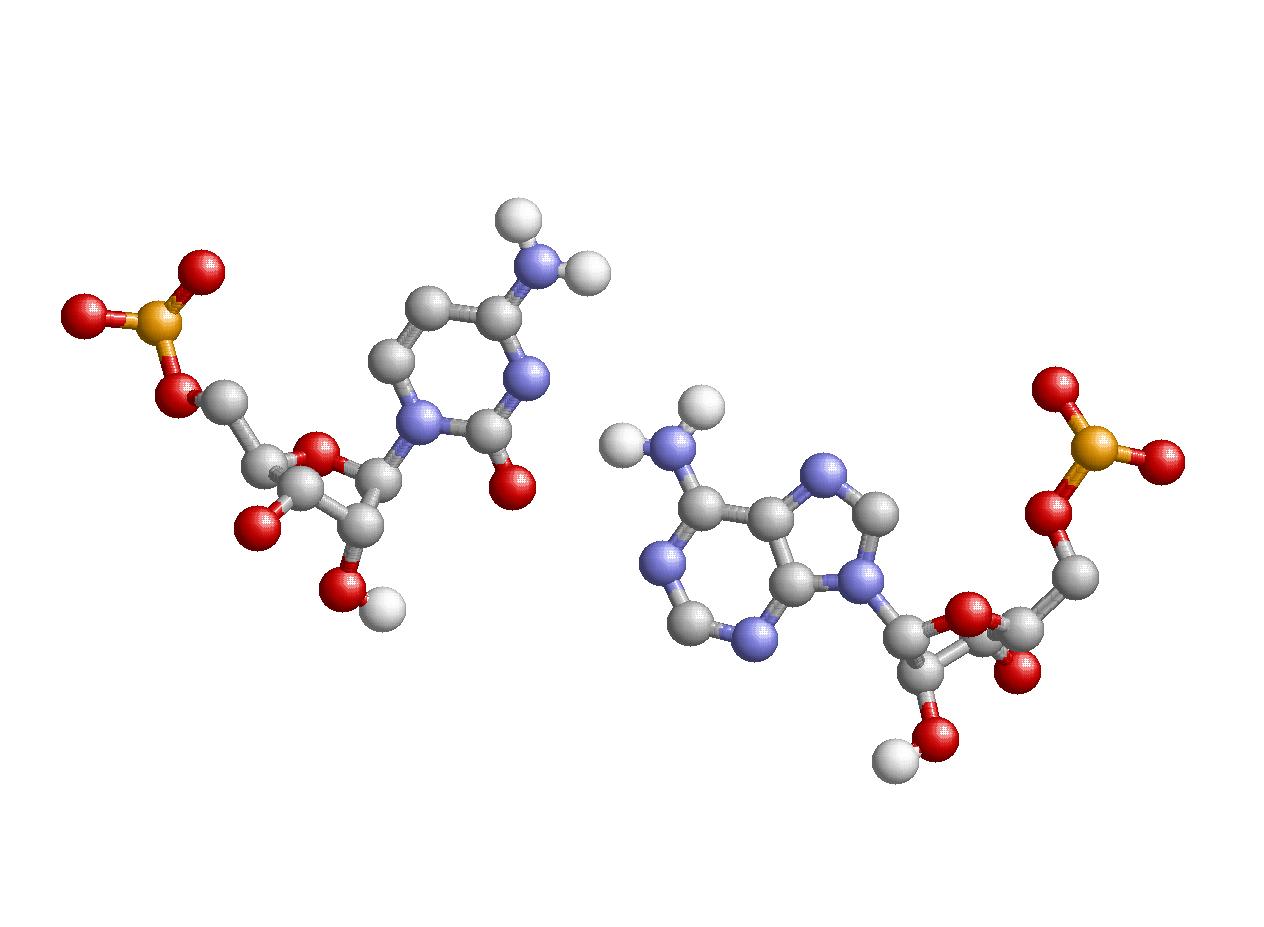
Структуру стеблевых дуплексов поддерживают 27 канонических и 5 неканонических пар оснований, в том числе и пару С-А, которая образована нуклеотидами 44 и 26, соответственно:
- Исследование третичной структуры
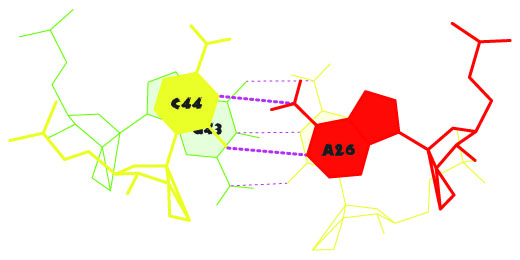
1. Данные о предположительных стрекинг-взаимодействиях получены в результате выполнения команды find pair -t 11QRT.pdb stdout | analyze и содержатся в выходном файле - 1QRT.out. Всего было выдано 28 потенциально возможных взаимодействий. Перекрытие GC/AC, обладающее наибольшей площадью (12,26 квадратных ангстрем), было замечено под номером 20.
Это - стрекинг-взаимодействие между основаниями G43 и C44 конца антикодонового стебля и C27 и А26 начала D-стебля.
Изображение соответствующего стекинг-взаимодействия получено при помощи выполнения последовательных команд:
ex_str -20 stacking.pdb step20.pdb (вырезание нужной структуры в отдельный файл),
stack2img –cdolt step20.pdb step20.ps (построение изображения).
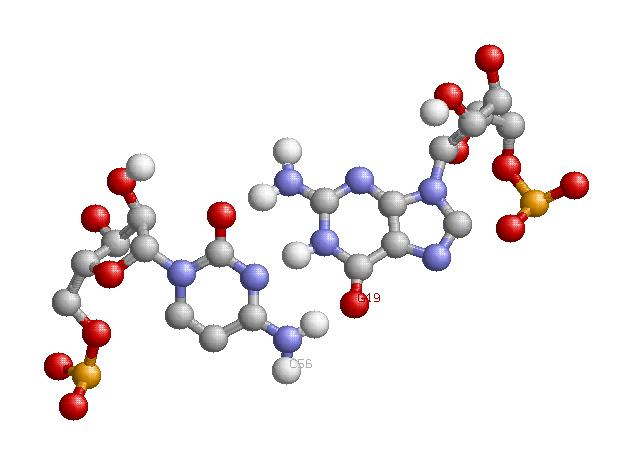
2. По данным файла 1QRT.fp существуют две дополнительные водородные связи между основаниями D- и Т-петель:
- Между G 19 и C 56, пара каноническая
- Между U 46 и U 47, пара неканоническая
Реальная и предсказанная вторичная структура тРНК из файла 1QRT.pdb
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' 2-7 3' 5' 66-71 3' Всего 6 пар |
Предсказано 6 пар из 6 реальных | Предсказано 6 пар из 6 реальных |
| D-стебель | 5’ 10-15 3’ 5’ 25-48 3’ Всего 6 пар |
Предсказано 0 пар из 6 реальных | Предсказано 3 пары из 6 реальных |
| T-стебель | 5’ 49-55 3’ 5’ 18-65 3’ Всего 7 пар |
Предсказано 0 пар из 6 реальных | Предсказано 5 пар из 7 реальных |
| Антикодоновый стебель | 5’ 37-44 3’ 5’ 26-33 3’ Всего 8 пар |
Предсказано 0 пар из 6 реальных | Предсказано 8 пар из 8 реальных |
| Общее число канонических пар нуклеотидов | 27 пар | 6 пар | 22 пары |
Предсказание с помощью einverted
Параметры программы:
Вводимая нуклеотидная последовательность:1QRT.fasta.txt
Штраф за гэп [12]: 12
Минимальное значение порога [50]:10 (при выборе 15, 20 или 50 программа выравнивание не находит)
Значение основания (канонической пары) [3]:3
Значение неканонической пары("несоответствие") [-4]:-4
Выходной файл – sequence.inv:
Программа рассчитана, по всей видимости, на работу с ДНК, а потому вместо U она выдает T. Если учесть это условие, можно заключить о предсказании всех 6 пар.
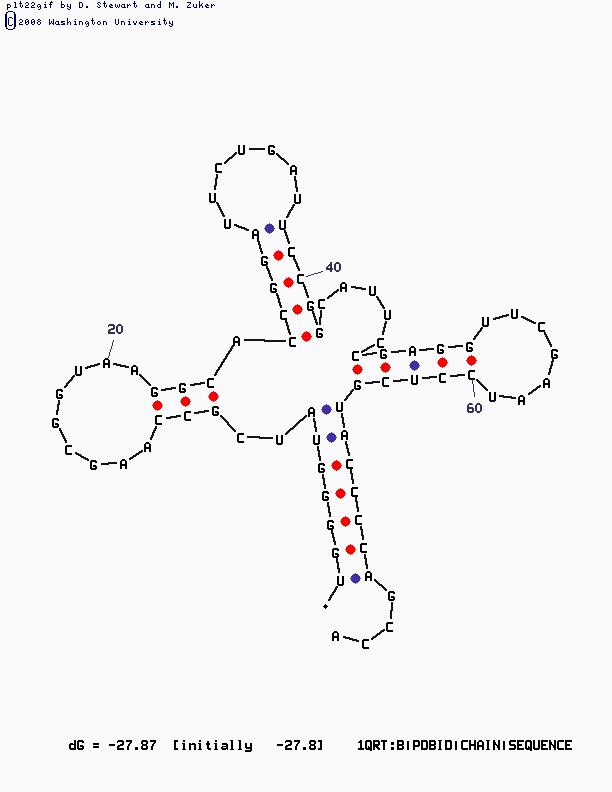
Алгоритм Зукера
Заданная команда, соответственно, со значением Р=15:
Mfold SEQ=1QRT.fasta P=15
Изображение, полученное с помощью mfold:
©Третьякова Светлана, 2008