» Семестры » Восьмой семестр » Молекулярная динамика биологических молекул в GROMACS
Молекулярная динамика биологических молекул в GROMACS¶
Целью данного занятия было ознакомиться с возможностями моделирования молекулярной динамики. Для этого использовался пакет молекулярной динамики Gromacs.
Моделирование самосборки липидного бислоя¶
Исходно были даны следующие файлы:
- дополнительной топологии для липида DPPC, dppc.itp.
- параметры для липидов lipid.itp.
- координаты одного липида dppc.gro.
- Файл-заготовка тополгии системы b.top.
- файл праметров для минимизации энергии em.mdp.
- файл праметров для "утряски" воды pr.mdp pr.mdp.
- файл праметров для молекулярной динамики md.mdp.
На основе одного липида создадим ячейку с 64 липидами:
%%bash
genconf -f dppc.gro -o b_64.gro -nbox 4 4 4
С помощью editconf преобразуем dppc.gro и b_64.gro в pdb-файлы:
%%bash
editconf -f dppc.gro -o dppc.pdb
editconf -f b_64.gro -o b_64.pdb
Ниже представлено графическое изображение. Видно, что b_64 - это 64 DPPC (оранжевый), которыми замостили пространство параллепипеда:
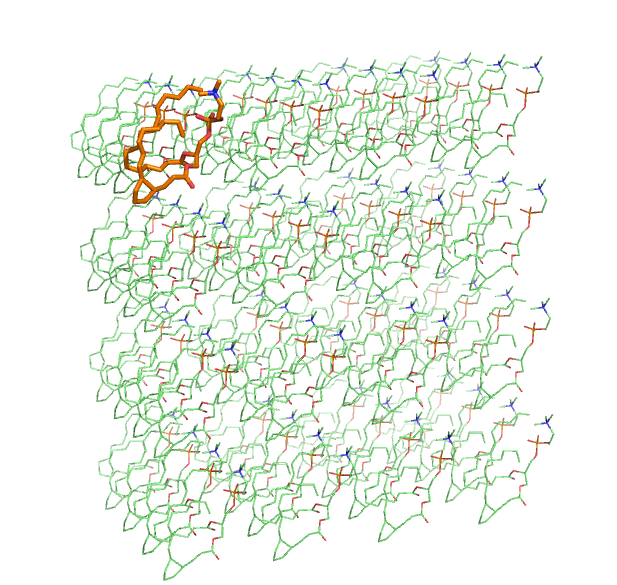
В текстовом редакторе в файле b.top установим правильное количество липидов в системе: DPPC 64.
Сделаем небольшой отступ в ячейке от липидов, что бы добавить примерно 2600 молекул воды:
%%bash
editconf -f b_64.gro -o b_ec -d 0.5
Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты молекул:
%%bash
grompp -f em -c b_ec -p b -o b_em -maxwarn 2
mdrun -deffnm b_em -v
Посмотрим изменение максимальной силы в ходе оптимизации геометрии. Начальное и конечное значение максимальной силы - 4.37970e+05 и 6.16887e+02. Видно, что сила резко уменьшилась.
%matplotlib inline
import numpy as np
import matplotlib.pyplot as plt
Fmax=[4.37970e+05, 3.57933e+04, 9.52568e+03, 1.50508e+04, 1.37059e+04, 4.02791e+03, 1.87751e+03,
3.76147e+03, 4.34365e+03, 1.91912e+03, 2.03533e+03, 3.00818e+03, 2.75324e+03, 8.43337e+02,
3.08327e+03, 2.09083e+03, 8.78173e+02, 1.07042e+03, 1.00741e+03, 1.77657e+03, 1.28547e+03,
6.94208e+02, 1.39504e+03, 1.44422e+03, 1.70904e+03, 3.29197e+02, 1.57250e+03, 8.45033e+02,
6.21543e+02, 6.19379e+02, 6.16887e+02]
steps = [0, 1, 2, 3, 4, 6, 9, 10, 11, 13, 15, 16, 17, 19, 20, 21, 23, 25, 26, 27, 28, 30, 31,
32, 33, 35, 36, 39, 42, 50, 51]
plt.plot(steps, Fmax, "m",alpha=0.5)
plt.ylabel('Fmax')
plt.xlabel('Step number')
plt.show()

Добавим в ячейку молекулы воды типа spc:
%%bash
genbox -cp b_em -p b -cs spc216 -o b_s
Проведём "утряску" воды:
%%bash
grompp -f pr -c b_s -p b -o b_pr -maxwarn 1
mdrun -deffnm b_pr -v
Переформатируем b_pr.gro и b_s.gro в pdb формат:
%%bash
editconf -f b_pr.gro -o b_pr.pdb
editconf -f b_s.gro -o b_s.pdb
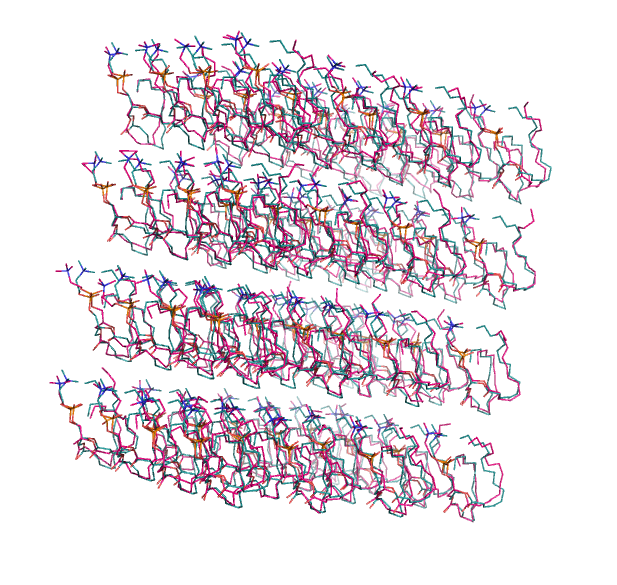
Теперь сравним их визуально в PyMol. Видно, что у DPPC различаются конформации в b_pr (розовый) и b_s (зелёный). Помимо этого поменялось и положение воды (не показано):
Последним шагом был запуск моделирования на суперкомпьютере:
%%bash
sbatch -N1 --ntasks-per-node=2 -e error-gpu.log -o output.log -t 350
-p gpu impi /opt/ccoe/gromacs-5.0.4/build/bin/gmx_mpi mdrun -testverlet -deffnm b_md -v
Дата последнего обновления: 20.05.17
© Светлана Яровенко