» Семестры » Восьмой семестр » Гомологичное моделирование комплекса белка с лигандом
Гомологичное моделирование комплекса белка с лигандом¶
Гомологическое моделирование проводилось для лизоцима из мотылька Hyalophora cecropia (Uniprot id: P05105 (LYS_HYACE)) на основе известной структуры лизоцима форели (PDB id: 1LMP).
Моделирование структуры белка производилось с помощью программы MODELLER, на вход которой подавался управляющий скрипт, файл pdb со структурой-образцом, файл выравнивания.
Сначала загружался модуль:
import sys
sys.path.append('/usr/lib/modeller9v7/modlib/')
sys.path.append('/usr/lib/modeller9v7/lib/x86_64-intel8/python2.5/')
import modeller
import _modeller
import modeller.automodel
Задавались некоторые начальные параметры:
env=modeller.environ()
env.io.hetatm=True
MODELLER 9v7, 2009/06/12, r6923
PROTEIN STRUCTURE MODELLING BY SATISFACTION OF SPATIAL RESTRAINTS
Copyright(c) 1989-2009 Andrej Sali
All Rights Reserved
Written by A. Sali
with help from
B. Webb, M.S. Madhusudhan, M-Y. Shen, M.A. Marti-Renom,
N. Eswar, F. Alber, M. Topf, B. Oliva, A. Fiser,
R. Sanchez, B. Yerkovich, A. Badretdinov,
F. Melo, J.P. Overington, E. Feyfant
University of California, San Francisco, USA
Rockefeller University, New York, USA
Harvard University, Cambridge, USA
Imperial Cancer Research Fund, London, UK
Birkbeck College, University of London, London, UK
Kind, OS, HostName, Kernel, Processor: 4, Linux kodomo.fbb.msu.ru 3.8-2-amd64 x86_64
Date and time of compilation : 2009/06/12 12:23:44
MODELLER executable type : x86_64-intel8
Job starting time (YY/MM/DD HH:MM:SS): 2017/05/03 18:21:52
Грузился белок-заготовка:
! wget http://www.pdb.org/pdb/files/1lmp.pdb
--2017-04-21 16:02:35-- http://www.pdb.org/pdb/files/1lmp.pdb
Resolving www.pdb.org... 132.249.231.77
Connecting to www.pdb.org|132.249.231.77|:80... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: http://www.rcsb.org/pdb/files/1lmp.pdb [following]
--2017-04-21 16:02:35-- http://www.rcsb.org/pdb/files/1lmp.pdb
Resolving www.rcsb.org... 132.249.231.10
Connecting to www.rcsb.org|132.249.231.10|:80... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: http://files.rcsb.org/view/1lmp.pdb [following]
--2017-04-21 16:02:35-- http://files.rcsb.org/view/1lmp.pdb
Resolving files.rcsb.org... 132.249.213.140
Connecting to files.rcsb.org|132.249.213.140|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: unspecified [text/plain]
Saving to: `1lmp.pdb.1'
[ <=> ] 131,301 160K/s in 0.8s
2017-04-21 16:02:37 (160 KB/s) - `1lmp.pdb.1' saved [131301]
И последовательность лизоцима из мотылька:
! wget http://www.uniprot.org/uniprot/P05105.fasta
--2017-04-21 16:03:26-- http://www.uniprot.org/uniprot/P05105.fasta Resolving www.uniprot.org... 128.175.240.211, 193.62.193.81 Connecting to www.uniprot.org|128.175.240.211|:80... connected. HTTP request sent, awaiting response... 200 OK Length: 205 [text/plain] Saving to: `P05105.fasta' 100%[======================================>] 205 --.-K/s in 0s 2017-04-21 16:03:27 (22.6 MB/s) - `P05105.fasta' saved [205/205]
Далее был создан объект - выравнивание, в которое были добавлены последовательности лизоцима из лосося и мотылька вместе со структурой первого.
alignm=modeller.alignment(env)
alignm.append(file='P05105.fasta', align_codes='all',alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
alignm[0].code="hyace"
alignm.salign()
alignm.write(file='all_in_one.ali', alignment_format='PIR')
read_pd_459W> Residue type NAG not recognized. 'automodel' model building
will treat this residue as a rigid body.
To use real parameters, add the residue type to ${LIB}/restyp.lib,
its topology to ${LIB}/top_*.lib, and suitable forcefield
parameters to ${LIB}/par.lib.
read_pd_459W> Residue type NDG not recognized. 'automodel' model building
will treat this residue as a rigid body.
To use real parameters, add the residue type to ${LIB}/restyp.lib,
its topology to ${LIB}/top_*.lib, and suitable forcefield
parameters to ${LIB}/par.lib.
SALIGN_____> adding the next group to the alignment; iteration 1
Далее строилась модель:
## Выбираем объект для моделирования
s = alignm[0]
pdb = alignm[1]
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='all_in_one.ali', knowns= pdb.code , sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 2
a.make()
hyace 1lmp
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
90 1 73 76 D C 8.895
END OF TABLE
read_to_681_> topology.submodel read from topology file: 3
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 139
atom names : C +N
atom indices : 1110 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 139
atom names : C CA +N O
atom indices : 1110 1107 0 1111
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 3
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
0 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
0 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 11694 10727
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_484W> Dihedral still outside +-90: -90.5221
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 139
Number of all, selected real atoms : 1112 1112
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 10727 10727
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2325
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1024.9258
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1136 0 1 0.007 0.007 18.182 1.000
2 Bond angle potential : 1532 1 9 2.183 2.183 146.36 1.000
3 Stereochemical cosine torsion poten: 734 0 28 46.896 46.896 248.18 1.000
4 Stereochemical improper torsion pot: 478 0 0 1.191 1.191 14.874 1.000
5 Soft-sphere overlap restraints : 2325 0 0 0.002 0.002 1.6931 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2217 0 2 0.349 0.349 101.37 1.000
10 Distance restraints 2 (N-O) : 2375 0 6 0.393 0.393 150.10 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 138 0 4 5.118 5.118 42.627 1.000
14 Sidechain Chi_1 dihedral restraints: 121 0 1 72.227 72.227 27.315 1.000
15 Sidechain Chi_2 dihedral restraints: 89 0 0 71.269 71.269 36.076 1.000
16 Sidechain Chi_3 dihedral restraints: 32 0 0 82.749 82.749 22.034 1.000
17 Sidechain Chi_4 dihedral restraints: 20 0 0 95.658 95.658 16.174 1.000
18 Disulfide distance restraints : 3 0 0 0.020 0.020 0.21478 1.000
19 Disulfide angle restraints : 6 0 0 3.256 3.256 1.4046 1.000
20 Disulfide dihedral angle restraints: 3 0 0 29.719 29.719 1.9015 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1240 0 0 0.460 0.460 25.831 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 137 24 21 33.303 71.622 144.90 1.000
26 Distance restraints 4 (SDCH-SDCH) : 466 0 1 0.639 0.639 25.697 1.000
27 Distance restraints 5 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: hyace.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 16088.8740
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2119 90D 90D N CA 713 714 124.20 107.00 17.20 4.95 107.00 17.20 4.95
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3909 17C 18D C N 131 133 -113.34 -63.30 70.86 11.78 -96.50 156.54 6.59
1 18D 18D N CA 133 134 -90.17 -40.00 114.20
2 3910 18D 19A C N 139 141 -80.17 -68.20 27.07 1.79 -62.50 150.56 25.58
2 19A 19A N CA 141 142 169.58 145.30 -40.90
3 3911 19A 20K C N 144 146 -70.18 -70.20 41.53 3.01 -62.90 137.47 18.26
3 20K 20K N CA 146 147 -178.08 140.40 -40.80
4 3912 20K 21R C N 153 155 -49.59 -72.10 24.46 2.05 -63.00 167.99 21.82
4 21R 21R N CA 155 156 151.45 141.90 -41.10
5 3913 21R 22F C N 164 166 -59.96 -71.40 11.70 0.72 -63.20 177.49 24.43
5 22F 22F N CA 166 167 138.24 140.70 -44.30
6 3914 22F 23T C N 175 177 -124.87 -124.80 1.90 0.08 -63.20 -173.22 28.04
6 23T 23T N CA 177 178 141.60 143.50 -42.10
7 3915 23T 24R C N 182 184 -85.56 -72.10 14.69 1.00 -63.00 172.61 24.87
7 24R 24R N CA 184 185 147.77 141.90 -41.10
8 3916 24R 25C C N 193 195 40.93 -69.10 133.37 12.31 -117.90 176.11 7.26
8 25C 25C N CA 195 196 -142.82 141.80 141.10
9 3918 26G 27L C N 203 205 -67.50 -70.70 21.08 1.48 -63.50 162.02 22.27
9 27L 27L N CA 205 206 120.77 141.60 -41.20
10 3919 27L 28V C N 211 213 -68.27 -62.40 7.35 0.89 -125.40 -172.37 10.44
10 28V 28V N CA 213 214 -37.97 -42.40 143.30
11 3920 28V 29Q C N 218 220 -133.53 -121.10 87.18 3.93 -63.80 116.81 20.63
11 29Q 29Q N CA 220 221 -134.01 139.70 -40.30
12 3921 29Q 30E C N 227 229 -58.61 -69.30 19.16 1.74 -63.60 161.37 21.25
12 30E 30E N CA 229 230 158.41 142.50 -40.30
13 3922 30E 31L C N 236 238 -72.28 -70.70 17.45 1.26 -63.50 160.07 22.74
13 31L 31L N CA 238 239 158.97 141.60 -41.20
14 3923 31L 32R C N 244 246 -70.86 -72.10 12.80 1.01 -63.00 164.45 22.75
14 32R 32R N CA 246 247 154.64 141.90 -41.10
15 3924 32R 33R C N 255 257 -96.50 -72.10 24.51 1.92 -63.00 -177.62 26.95
15 33R 33R N CA 257 258 139.62 141.90 -41.10
16 3925 33R 34L C N 266 268 -106.57 -108.50 16.51 0.87 -63.50 163.09 20.61
16 34L 34L N CA 268 269 116.10 132.50 -41.20
17 3926 34L 35G C N 274 276 84.00 78.70 51.72 1.26 82.20 133.97 6.59
17 35G 35G N CA 276 277 142.46 -166.10 8.50
18 3929 37D 38E C N 297 299 64.65 54.60 13.27 0.88 -63.60 148.08 25.48
18 38E 38E N CA 299 300 33.73 42.40 -40.30
19 3943 51E 52S C N 411 413 -138.91 -64.10 82.71 8.71 -64.10 82.71 8.71
19 52S 52S N CA 413 414 0.29 -35.00 -35.00
20 3952 60G 61K C N 479 481 -98.90 -118.00 92.20 4.18 -70.20 95.89 7.57
20 61K 61K N CA 481 482 48.90 139.10 140.40
21 3974 82S 83K C N 664 666 -73.41 -62.90 94.82 12.91 -62.90 94.82 12.91
21 83K 83K N CA 666 667 -135.03 -40.80 -40.80
22 3975 83K 84G C N 673 675 -102.14 -62.40 39.85 7.23 82.20 -176.64 10.01
22 84G 84G N CA 675 676 -44.11 -41.20 8.50
23 3981 89K 90D C N 711 713 -129.62 -63.30 75.38 12.87 -63.30 75.38 12.87
23 90D 90D N CA 713 714 -75.82 -40.00 -40.00
24 4005 113I 114Y C N 884 886 -53.19 -98.40 46.16 2.11 -63.50 162.80 26.36
24 114Y 114Y N CA 886 887 119.07 128.40 -43.40
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 6 10 69 69 122 114 132 151 173 185
<< end of ENERGY.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 139
Number of all, selected real atoms : 1112 1112
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 10727 10727
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2364
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 937.2785
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1136 0 1 0.008 0.008 18.493 1.000
2 Bond angle potential : 1532 1 7 2.052 2.052 130.91 1.000
3 Stereochemical cosine torsion poten: 734 0 19 45.819 45.819 241.14 1.000
4 Stereochemical improper torsion pot: 478 0 0 1.186 1.186 14.924 1.000
5 Soft-sphere overlap restraints : 2364 0 0 0.002 0.002 1.6831 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2217 0 3 0.398 0.398 112.65 1.000
10 Distance restraints 2 (N-O) : 2375 0 7 0.438 0.438 156.91 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 138 0 3 4.915 4.915 39.322 1.000
14 Sidechain Chi_1 dihedral restraints: 121 0 1 71.365 71.365 35.536 1.000
15 Sidechain Chi_2 dihedral restraints: 89 0 0 65.964 65.964 31.572 1.000
16 Sidechain Chi_3 dihedral restraints: 32 0 0 88.883 88.883 23.176 1.000
17 Sidechain Chi_4 dihedral restraints: 20 0 0 97.787 97.787 14.187 1.000
18 Disulfide distance restraints : 3 0 0 0.023 0.023 0.28267 1.000
19 Disulfide angle restraints : 6 0 0 2.174 2.174 0.62624 1.000
20 Disulfide dihedral angle restraints: 3 0 0 32.706 32.706 2.6644 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1240 0 0 0.448 0.448 23.750 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 137 19 14 27.428 65.588 63.675 1.000
26 Distance restraints 4 (SDCH-SDCH) : 466 0 0 0.646 0.646 25.777 1.000
27 Distance restraints 5 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: hyace.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 15443.5273
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2119 90D 90D N CA 713 714 123.60 107.00 16.60 4.77 107.00 16.60 4.77
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3910 18D 19A C N 139 141 -76.50 -68.20 8.66 0.65 -62.50 171.91 28.88
1 19A 19A N CA 141 142 147.76 145.30 -40.90
2 3912 20K 21R C N 153 155 -79.17 -72.10 7.85 0.53 -63.00 174.33 24.65
2 21R 21R N CA 155 156 145.33 141.90 -41.10
3 3913 21R 22F C N 164 166 -81.32 -71.40 13.39 1.17 -63.20 176.94 23.37
3 22F 22F N CA 166 167 131.71 140.70 -44.30
4 3914 22F 23T C N 175 177 -72.11 -78.10 24.53 0.87 -63.20 168.35 21.01
4 23T 23T N CA 177 178 126.01 149.80 -42.10
5 3915 23T 24R C N 182 184 -96.38 -72.10 30.50 1.99 -63.00 162.03 24.19
5 24R 24R N CA 184 185 160.35 141.90 -41.10
6 3917 25C 26G C N 199 201 170.96 -167.20 22.16 0.35 82.20 -168.35 13.32
6 26G 26G N CA 201 202 178.36 174.60 8.50
7 3918 26G 27L C N 203 205 -75.40 -70.70 10.90 0.68 -63.50 167.79 24.00
7 27L 27L N CA 205 206 151.43 141.60 -41.20
8 3920 28V 29Q C N 218 220 -92.30 -73.00 34.69 1.99 -63.80 152.85 24.00
8 29Q 29Q N CA 220 221 169.53 140.70 -40.30
9 3922 30E 31L C N 236 238 -83.37 -70.70 12.76 1.20 -63.50 179.76 26.14
9 31L 31L N CA 238 239 140.14 141.60 -41.20
10 3923 31L 32R C N 244 246 -139.43 -125.20 14.39 0.52 -63.00 -168.01 31.17
10 32R 32R N CA 246 247 142.78 140.60 -41.10
11 3924 32R 33R C N 255 257 -127.64 -125.20 45.83 2.27 -63.00 150.52 17.16
11 33R 33R N CA 257 258 94.83 140.60 -41.10
12 3925 33R 34L C N 266 268 -136.07 -108.50 29.66 1.32 -63.50 -170.22 30.84
12 34L 34L N CA 268 269 143.45 132.50 -41.20
13 3926 34L 35G C N 274 276 62.21 78.70 22.40 0.54 82.20 160.70 8.80
13 35G 35G N CA 276 277 -150.95 -166.10 8.50
14 3928 36F 37D C N 289 291 -47.60 -63.30 15.70 2.48 54.50 130.44 16.66
14 37D 37D N CA 291 292 -40.28 -40.00 40.90
15 3943 51E 52S C N 411 413 -138.23 -64.10 82.32 8.62 -64.10 82.32 8.62
15 52S 52S N CA 413 414 0.78 -35.00 -35.00
16 3952 60G 61K C N 479 481 -100.73 -118.00 79.39 3.59 -70.20 84.50 6.77
16 61K 61K N CA 481 482 61.61 139.10 140.40
17 3953 61K 62V C N 488 490 -65.30 -62.40 7.82 0.87 -125.40 -171.72 7.63
17 62V 62V N CA 490 491 -35.13 -42.40 143.30
18 3981 89K 90D C N 711 713 -138.30 -63.30 83.50 14.25 -63.30 83.50 14.25
18 90D 90D N CA 713 714 -76.69 -40.00 -40.00
19 4005 113I 114Y C N 884 886 -49.54 -98.40 49.54 2.17 -63.50 164.22 26.78
19 114Y 114Y N CA 886 887 120.22 128.40 -43.40
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 8 18 57 79 129 99 137 169 153 208
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
hyace.B99990001.pdb 1024.92578
hyace.B99990002.pdb 937.27850
import nglview
import ipywidgets
w1 = nglview.show_structure_file('hyace.B99990001.pdb')
w1
Изображение полученной структуры:
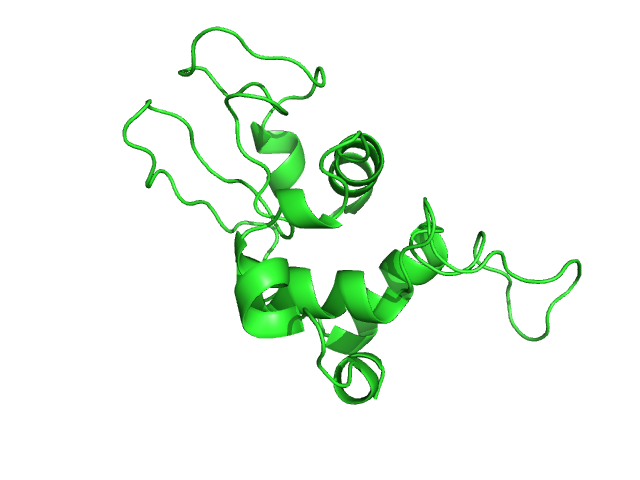
Видно, что в полученной структуре отсутсвуют лиганды (нет NAG и NDG), которые есть в лизоциме форели.
Добавим их:
res_list1 = [res.code for res in alignm[0].residues]
new_seq = "".join(res_list1)+"..."
alignm=modeller.alignment(env)
alignm.append(file='P05105.fasta', align_codes='all',alignment_format='FASTA')
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
alignm[0].code="hyace"
alignm.append_sequence(new_seq)
alignm[2].code="hyace_lig"
alignm.salign()
alignm.write(file='all_in_one.ali', alignment_format='PIR')
SALIGN_____> adding the next group to the alignment; iteration 1 SALIGN_____> adding the next group to the alignment; iteration 2
## Выбираем объект для моделирования
with_lig = alignm[2]
pdb = alignm[1]
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='all_in_one.ali', knowns= pdb.code , sequence = with_lig.code )
a.name='mod'+with_lig.code
a.starting_model = 1
a.ending_model = 2
a.make()
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
90 1 73 76 D C 8.895
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 139
atom names : C +N
atom indices : 1110 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 139
atom names : C CA +N O
atom indices : 1110 1107 0 1111
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 3
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 13083 12116
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 142
Number of all, selected real atoms : 1155 1155
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 12116 12116
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2582
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1166.6425
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1136 0 1 0.009 0.009 26.101 1.000
2 Bond angle potential : 1532 1 12 2.358 2.358 171.26 1.000
3 Stereochemical cosine torsion poten: 734 0 29 46.674 46.674 249.84 1.000
4 Stereochemical improper torsion pot: 478 0 0 1.455 1.455 21.540 1.000
5 Soft-sphere overlap restraints : 2582 1 2 0.007 0.007 16.440 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2217 0 5 0.370 0.370 114.74 1.000
10 Distance restraints 2 (N-O) : 2375 0 14 0.492 0.492 210.15 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 138 0 2 5.037 5.037 41.291 1.000
14 Sidechain Chi_1 dihedral restraints: 121 0 2 74.927 74.927 41.494 1.000
15 Sidechain Chi_2 dihedral restraints: 89 0 2 65.225 65.225 37.688 1.000
16 Sidechain Chi_3 dihedral restraints: 32 0 0 77.785 77.785 18.116 1.000
17 Sidechain Chi_4 dihedral restraints: 20 0 0 90.820 90.820 12.417 1.000
18 Disulfide distance restraints : 3 0 0 0.023 0.023 0.27912 1.000
19 Disulfide angle restraints : 6 0 0 3.765 3.765 1.8777 1.000
20 Disulfide dihedral angle restraints: 3 0 0 20.233 20.233 1.2433 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1240 0 0 0.376 0.376 19.154 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 137 23 29 35.932 71.419 122.16 1.000
26 Distance restraints 4 (SDCH-SDCH) : 466 0 1 0.751 0.751 27.533 1.000
27 Distance restraints 5 (X-Y) : 1389 0 2 0.055 0.055 33.315 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: hyace_lig.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 16913.0332
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2119 90D 90D N CA 713 714 124.71 107.00 17.72 5.09 107.00 17.72 5.09
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3910 18D 19A C N 139 141 -69.08 -68.20 11.93 0.93 -62.50 162.03 26.88
1 19A 19A N CA 141 142 157.20 145.30 -40.90
2 3911 19A 20K C N 144 146 -73.06 -70.20 5.91 0.36 -62.90 173.93 23.16
2 20K 20K N CA 146 147 145.57 140.40 -40.80
3 3912 20K 21R C N 153 155 -63.67 -72.10 12.24 1.06 -63.00 168.14 22.76
3 21R 21R N CA 155 156 150.76 141.90 -41.10
4 3913 21R 22F C N 164 166 -66.50 -71.40 11.91 0.80 -63.20 174.18 23.96
4 22F 22F N CA 166 167 129.85 140.70 -44.30
5 3914 22F 23T C N 175 177 -126.41 -124.80 50.56 2.12 -63.20 139.06 21.93
5 23T 23T N CA 177 178 -165.96 143.50 -42.10
6 3915 23T 24R C N 182 184 -59.22 -72.10 13.98 0.95 -63.00 177.60 24.25
6 24R 24R N CA 184 185 136.46 141.90 -41.10
7 3917 25C 26G C N 199 201 -95.87 -80.20 59.25 1.61 82.20 -135.17 17.52
7 26G 26G N CA 201 202 -128.76 174.10 8.50
8 3918 26G 27L C N 203 205 -64.01 -70.70 7.16 0.55 -63.50 179.76 24.99
8 27L 27L N CA 205 206 139.04 141.60 -41.20
9 3920 28V 29Q C N 218 220 -64.57 -73.00 30.22 1.82 -63.80 151.98 22.31
9 29Q 29Q N CA 220 221 111.68 140.70 -40.30
10 3922 30E 31L C N 236 238 -122.90 -108.50 39.61 1.96 -63.50 160.77 26.00
10 31L 31L N CA 238 239 169.40 132.50 -41.20
11 3923 31L 32R C N 244 246 -131.77 -125.20 8.27 0.44 -63.00 -170.42 21.94
11 32R 32R N CA 246 247 135.57 140.60 -41.10
12 3925 33R 34L C N 266 268 70.89 -70.70 148.15 11.86 -63.50 -166.51 34.75
12 34L 34L N CA 268 269 98.00 141.60 -41.20
13 3926 34L 35G C N 274 276 89.37 78.70 53.76 1.11 82.20 132.90 6.81
13 35G 35G N CA 276 277 141.21 -166.10 8.50
14 3928 36F 37D C N 289 291 48.70 -70.90 134.74 11.31 54.50 171.57 12.15
14 37D 37D N CA 291 292 -147.63 150.30 40.90
15 3929 37D 38E C N 297 299 -114.93 -117.80 84.23 4.18 -63.60 106.16 12.28
15 38E 38E N CA 299 300 52.62 136.80 -40.30
16 3956 64K 65N C N 512 514 -82.11 -119.90 54.80 2.97 -63.20 143.47 19.00
16 65N 65N N CA 514 515 176.69 137.00 -41.10
17 3957 65N 66G C N 520 522 -62.71 -62.40 20.43 3.04 82.20 147.84 10.83
17 66G 66G N CA 522 523 -20.78 -41.20 8.50
18 3978 86T 87P C N 691 693 -55.13 -58.70 46.24 3.54 -64.50 136.53 10.69
18 87P 87P N CA 693 694 -76.60 -30.50 147.20
19 3979 87P 88G C N 698 700 -65.69 -62.40 11.33 1.87 82.20 159.81 12.29
19 88G 88G N CA 700 701 -52.04 -41.20 8.50
20 3980 88G 89K C N 702 704 60.17 -70.20 130.47 9.64 -70.20 130.47 9.64
20 89K 89K N CA 704 705 145.35 140.40 140.40
21 3981 89K 90D C N 711 713 -152.18 -63.30 94.27 15.98 -63.30 94.27 15.98
21 90D 90D N CA 713 714 -71.43 -40.00 -40.00
22 4005 113I 114Y C N 884 886 -54.27 -98.40 49.97 3.18 -63.50 148.65 24.06
22 114Y 114Y N CA 886 887 104.96 128.40 -43.40
23 4029 137S 138D C N 1096 1098 -95.49 -70.90 72.17 2.54 -63.30 106.82 15.26
23 138D 138D N CA 1098 1099 -141.85 150.30 -40.00
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 11 12 81 108 121 122 153 170 183 205
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 142
Number of all, selected real atoms : 1155 1155
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 12116 12116
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2446
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1100.8982
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1136 0 1 0.008 0.008 23.307 1.000
2 Bond angle potential : 1532 3 10 3.037 3.037 186.60 1.000
3 Stereochemical cosine torsion poten: 734 0 31 48.563 48.563 266.94 1.000
4 Stereochemical improper torsion pot: 478 0 0 1.293 1.293 17.735 1.000
5 Soft-sphere overlap restraints : 2446 1 2 0.007 0.007 16.176 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2217 0 2 0.330 0.330 95.315 1.000
10 Distance restraints 2 (N-O) : 2375 0 7 0.388 0.388 146.22 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 138 0 2 4.808 4.808 37.617 1.000
14 Sidechain Chi_1 dihedral restraints: 121 0 1 76.086 76.086 36.961 1.000
15 Sidechain Chi_2 dihedral restraints: 89 0 0 76.633 76.633 41.569 1.000
16 Sidechain Chi_3 dihedral restraints: 32 0 0 91.689 91.689 22.779 1.000
17 Sidechain Chi_4 dihedral restraints: 20 0 0 84.121 84.121 13.236 1.000
18 Disulfide distance restraints : 3 0 0 0.025 0.025 0.32193 1.000
19 Disulfide angle restraints : 6 0 0 2.614 2.614 0.90503 1.000
20 Disulfide dihedral angle restraints: 3 0 0 24.182 24.182 1.8126 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1240 0 0 0.446 0.446 31.741 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 137 22 19 30.941 72.235 94.477 1.000
26 Distance restraints 4 (SDCH-SDCH) : 466 0 0 0.807 0.807 34.396 1.000
27 Distance restraints 5 (X-Y) : 1389 0 2 0.054 0.054 32.791 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: hyace_lig.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 17260.0801
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2119 90D 90D N CA 713 714 125.57 107.00 18.57 5.34 107.00 18.57 5.34
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3910 18D 19A C N 139 141 178.81 -134.00 53.26 1.22 -62.50 -170.74 37.21
1 19A 19A N CA 141 142 171.69 147.00 -40.90
2 3911 19A 20K C N 144 146 -111.89 -118.00 19.48 0.85 -62.90 168.67 19.79
2 20K 20K N CA 146 147 120.60 139.10 -40.80
3 3912 20K 21R C N 153 155 -150.36 -125.20 25.70 1.17 -63.00 -163.08 22.40
3 21R 21R N CA 155 156 135.38 140.60 -41.10
4 3913 21R 22F C N 164 166 -147.80 -124.20 23.87 1.04 -63.20 -164.73 32.57
4 22F 22F N CA 166 167 139.71 143.30 -44.30
5 3914 22F 23T C N 175 177 -100.68 -78.10 23.13 1.26 -63.20 167.33 23.96
5 23T 23T N CA 177 178 154.83 149.80 -42.10
6 3915 23T 24R C N 182 184 -123.75 -125.20 23.79 1.12 -63.00 169.23 19.61
6 24R 24R N CA 184 185 116.86 140.60 -41.10
7 3917 25C 26G C N 199 201 -75.73 -80.20 5.94 0.43 82.20 -128.33 6.98
7 26G 26G N CA 201 202 178.00 174.10 8.50
8 3918 26G 27L C N 203 205 -68.40 -70.70 7.73 0.67 -63.50 169.89 23.87
8 27L 27L N CA 205 206 148.98 141.60 -41.20
9 3919 27L 28V C N 211 213 -62.56 -62.40 1.75 0.23 -125.40 -176.36 10.30
9 28V 28V N CA 213 214 -44.14 -42.40 143.30
10 3921 29Q 30E C N 227 229 95.12 -117.80 148.59 5.98 -63.60 -137.50 37.75
10 30E 30E N CA 229 230 115.63 136.80 -40.30
11 3923 31L 32R C N 244 246 -18.71 -72.10 55.89 3.91 -63.00 172.26 26.33
11 32R 32R N CA 246 247 125.37 141.90 -41.10
12 3924 32R 33R C N 255 257 -150.41 -125.20 25.26 0.97 -63.00 -162.91 32.57
12 33R 33R N CA 257 258 142.25 140.60 -41.10
13 3925 33R 34L C N 266 268 -119.61 -108.50 20.30 0.95 -63.50 178.36 28.23
13 34L 34L N CA 268 269 149.49 132.50 -41.20
14 3926 34L 35G C N 274 276 100.02 78.70 37.42 0.62 82.20 155.67 8.44
14 35G 35G N CA 276 277 163.15 -166.10 8.50
15 3929 37D 38E C N 297 299 57.44 54.60 4.03 0.25 -63.60 145.00 24.94
15 38E 38E N CA 299 300 39.54 42.40 -40.30
16 3952 60G 61K C N 479 481 -106.29 -118.00 81.68 3.79 -70.20 89.71 7.25
16 61K 61K N CA 481 482 58.27 139.10 140.40
17 3981 89K 90D C N 711 713 -143.80 -63.30 87.85 14.97 -63.30 87.85 14.97
17 90D 90D N CA 713 714 -75.16 -40.00 -40.00
18 4005 113I 114Y C N 884 886 -48.58 -98.40 54.65 3.24 -63.50 150.08 24.58
18 114Y 114Y N CA 886 887 105.93 128.40 -43.40
19 4008 116R 117H C N 916 918 -97.82 -125.60 50.67 1.36 -63.20 142.98 16.02
19 117H 117H N CA 918 919 96.42 138.80 -42.30
20 4009 117H 118K C N 926 928 -55.06 -62.90 70.41 8.74 -118.00 126.84 6.57
20 118K 118K N CA 928 929 -110.78 -40.80 139.10
21 4019 127N 128H C N 1020 1022 -121.22 -63.20 74.38 8.76 -63.20 74.38 8.76
21 128H 128H N CA 1022 1023 4.24 -42.30 -42.30
22 4020 128H 129C C N 1030 1032 -126.56 -63.00 65.20 10.00 -63.00 65.20 10.00
22 129C 129C N CA 1032 1033 -26.57 -41.10 -41.10
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 10 19 65 77 131 134 139 167 164 179
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
hyace_lig.B99990001.pdb 1166.64246
hyace_lig.B99990002.pdb 1100.89819
import nglview
import ipywidgets
w3 = nglview.show_structure_file('hyace_lig.B99990001.pdb')
w3
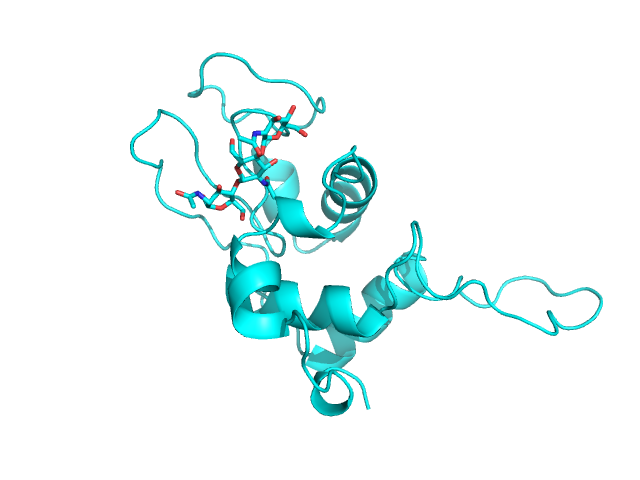
Изображение полученной структуры уже с лигандами:
А здесь показано наложение полученной модели (синий цвет) и структуры лизоцима форели (розовый), полученное командой align в PyMOL:
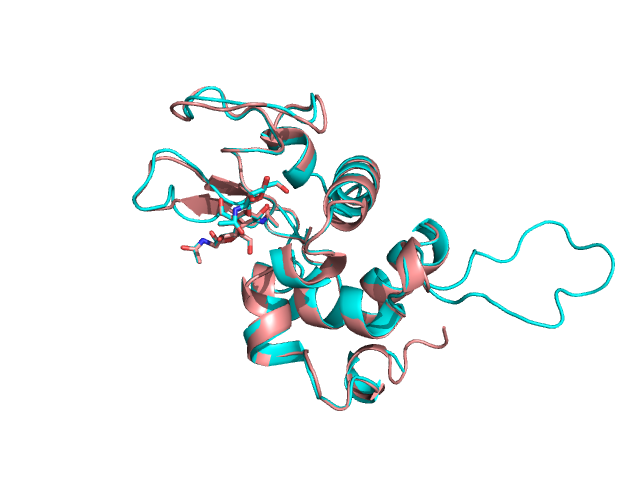
Видно, что у модели есть участок с большим выпетливанием, а также, что рядом с лигандами находится небольшой β-лист, где у белка-затравки расположена петля.
Далее была предпринята попытка перенести один из лигандов на конец белка, но она не удалась, несмотря на то, что координаты соответствовали тем, что находятся в pdb-файле.
class MyModel(modeller.automodel.automodel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('CA:129', 'O:132'),
('O:129', 'N2:132'),
('N:129', 'O3:132')):
atoms = [self.atoms[i] for i in ids]
#rsr.add(forms.upper_bound(group=physical.upper_distance,
# feature=features.distance(*atoms), mean=3.5, stdev=0.1))
env.libs.topology.clear()
a = MyModel(env, alnfile='all_in_one.ali', knowns= "1lmp" , sequence = 'hyace_lig')
a.name='mod'+"try1"
a.starting_model = 1
a.ending_model = 2
a.make()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
90 1 73 76 D C 8.895
END OF TABLE
read_to_681_> topology.submodel read from topology file: 3
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 139
atom names : C +N
atom indices : 1110 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 139
atom names : C CA +N O
atom indices : 1110 1107 0 1111
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 3
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
--------------------------------------------------------------------------- KeyError Traceback (most recent call last) <ipython-input-79-aafcfc4bd2d2> in <module>() 2 a.starting_model = 1 3 a.ending_model = 2 ----> 4 a.make() /usr/lib/modeller9v7/modlib/modeller/automodel/automodel.py in make(self, exit_stage) 96 97 self.outputs = [] ---> 98 self.homcsr(exit_stage) 99 # Exit early? 100 if exit_stage >= 1: /usr/lib/modeller9v7/modlib/modeller/automodel/automodel.py in homcsr(self, exit_stage) 433 # make and write the stereochemical, homology, and special restraints? 434 if self.create_restraints: --> 435 self.mkhomcsr(selection(self), aln) 436 self.restraints.condense() 437 self.restraints.write(self.csrfile) /usr/lib/modeller9v7/modlib/modeller/automodel/automodel.py in mkhomcsr(self, atmsel, aln) 521 # Special restraints have to be called last so that possible cis-proline 522 # changes are reflected in the current restraints: --> 523 self.special_restraints(aln) 524 525 def distance_restraints(self, atmsel, aln): <ipython-input-78-e64d7ba7d1ea> in special_restraints(self, aln) 5 ('O:129', 'N2:132'), 6 ('N:129', 'O3:132')): ----> 7 atoms = [self.atoms[i] for i in ids] 8 #rsr.add(forms.upper_bound(group=physical.upper_distance, 9 # feature=features.distance(*atoms), mean=3.5, stdev=0.1)) /usr/lib/modeller9v7/modlib/modeller/coordinates.py in __getitem__(self, indx) 359 def __getitem__(self, indx): 360 ret = modutil.handle_seq_indx(self, indx, self.mdl._indxatm, --> 361 (self.offset, self.length, self.suffix)) 362 if isinstance(ret, int): 363 return self.mdl._atom_class(self.mdl, ret + self.offset) /usr/lib/modeller9v7/modlib/modeller/util/modutil.py in handle_seq_indx(seqtype, indx, lookup_func, lookup_args, require_inrange) 17 elif lookup_func is not None: 18 args = lookup_args + (indx,) ---> 19 int_indx = lookup_func(*args) 20 if int_indx < 0: 21 raise KeyError(indx) /usr/lib/modeller9v7/modlib/modeller/coordinates.py in _indxatm(self, offset, length, suffix, indx) 272 newindx = func(self.cdpt, self.seqpt, indx+suffix) - 1 - offset 273 if newindx < 0 or (length is not None and newindx >= length): --> 274 raise KeyError("No such atom: %s" % indx) 275 return newindx 276 raise TypeError("Atom IDs must be numbers or strings") KeyError: 'No such atom: N2:132'
Дата последнего обновления: 04.05.17
© Светлана Яровенко