BIOB_gene → mutant1 → mutant2 → mutant3 → mutant4 → mutant5 → mutant6
Модель создается в предположении, что на каждом следующем этапе в последовательности нашего гена происходит определенное число замен, в соответствии с таблицей| замен на 100 нуклеотидов | замен на всю последовательность (1041 нуклеотид) | |
| mut1 | 10 | 104 |
| mut2 | 10 | 104 |
| mut3 | 30 | 312 |
| mut4 | 25 | 260 |
| mut5 | 50 | 520 |
| mut6 | 50 | 520 |
Мутантные последовательности получали с помощью программы msbar из пакета EMBOSS. Общий вид команды UNIX выглядит следующим образом.
msbar "входной файл" "исходящий файл" -point 4 -count "общее количество замен" -auto
Параметр -point 4 означает, что программа будет делать в последовательности только замены. Для получения сразу всех мутантных последовательностей в одном файле был написан скрипт, текст которого можно посмотреть script1.chmod. В результате выполнения данного скрипта был получен файл, содержащий все мутантные последовательности. Все дальнейшие операции (исследования) проводились с этим файлом.
Определяем попарные эволюционные расстояния между всеми последовательностями c помощью программы distmat пакета EMBOSS. Устанавливая параметр -nucmethod равным 0 и 1, мы получим матрицу попарных различий и матрицу попарных расстояний, вычисленных по формуле Джукса – Кантора соответственно. Синтаксис команды выглядит следующим образом:
distmat -sequence "входящий файл" -outfile "исходящий файл" -nucmethod "метод оценки (0 или 1)"
Исходя из полученных данных составили сводный файл Excel. В этом файле мы пытались оценить насколько результаты определения попарных эволюционных расстояний отличаются от истинных. Оказалось, что отличаются достаточно сильно. Эволюционные расстояния, вычисленные по формуле Джукса – Кантора ближе к истинным, чем попарные различия (несовпадения). Рассматривая график зависимости истинных расстояний от эволюционных расстояний вычисленных двумя разными способами можно заметить, что вычисленные расстояния близки к истинным при малых количествах эволюцонных событий.
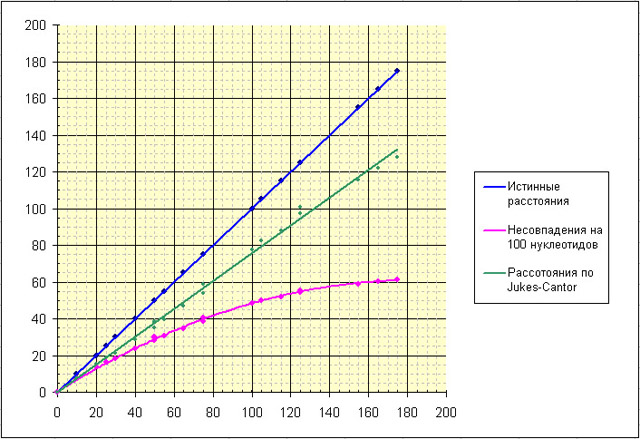
Это вполне логично, поскольку чем меньше замен произошло, тем меньше вероятность, что в одном нуклеотиде произойдет более одной замены. Таким образом, начиная приблизительно с 50 замен на 100 нуклеотидов отклонение оценки от истинного значения становится существенно. Основной вывод - чем меньше замен, тем точнее оценка!
Примечание: На графике точки соединены линиями тренда, для графиков истинных значений и расстояний по Джуксу-Кантору линия тренда - линейная функция; график несовпадений на 100 нуклеотидов - график
полинома 2-ой степени.