NGS: качество чтений, фильтрация
1. Описание файла с референсом
Референсный геном находится в файле chr6.fna и представляет собой по сути обычный fasta-файл с единственной последовательностью. Идентифекатор последовательности - NC_000006.12 - является одновременно AC этой последовательности в базе данных nuccore. В описании последовательности указана прочая основная информация: "Homo sapiens chromosome 6, GRCh38.p13 Primary Assembly", то есть организм, хромосома, название сборки.
Также можно видеть, что начало и конец последовательности целиком состоят и неизвестных нуклеотидов (N).
2. Индексация референса
Индексация выполнена командой
bwa index -a bwtsw chr6.fna &> index.logВерсия bwa 0.7.17-r1188, log-файл
Параметр -a выбирает алгоритм, в данном случае -- bwtsw. Второй существующий алгоритм "is" быстрее, но не работает с чем-то длиннее 2GB (хотя в моей хромосоме 170 805 979 пн, т.е. меньше одного gb, так что можно было бы использовать и is). Также существует опциональный параметр -p, которым можно задать название проиндесированной базы, иначе будет использовано название исходного файла.
На вход был подан референсный геном, на выходе получилось пять файлов:
- chr6.fna.amb - содержит индексы всех неоднозначностей: в первой строке общая длина последовательности, число последовательностей в файле и число оставшихся строк в файле, т.е. число блоков из стоящих подряд неоднозначных нуклеотидов определённого типа. Каждая последующая строка содержит информацию об одном таком блоке: начало, длину и тип нуклеотида (в моём случае N или Y).
- chr6.fna.ann - короткий файл с общей информацией: в первой строке суммарная длина последовательностей в файле, число последовательностей и непонятное 11, потом пары строк: 0, название последовательности и её описание, а в следующей строке индекс начала, длина и число блоков неоднозначностей в ней.
- Остальные три файла - chr6.fna.btw, chr6.fna.pac, chr6.fna.sa являются бинарными и чтению не поддаются, но они должны содержать индексы различных n-грамм и какую-то дополнительную внутреннюю информацию для дальнейшей работы.
3. Описание образца
| ссылка | https://www.ncbi.nlm.nih.gov/sra/SRR10720416 |
|---|---|
| прибор | Illumina Genome Analyzer IIx |
| организм | Homo sapiens |
| стратегия секвенирования | other |
| чтения | парные |
| чтений ожидается (spots) | 41M |
4. Проверка качества исходных чтений
fastqc -o fastqc_analysis --extract SRR10720416_?.fastq.gz &> fastqc_analysis/fastqc.log
Версия FastQC v0.11.8, log-файл.
Прямых и обратных чтений одинаковое число: по 40 990 418 и это совпадает с предварительной оценкой (41М).
Per base sequence quality
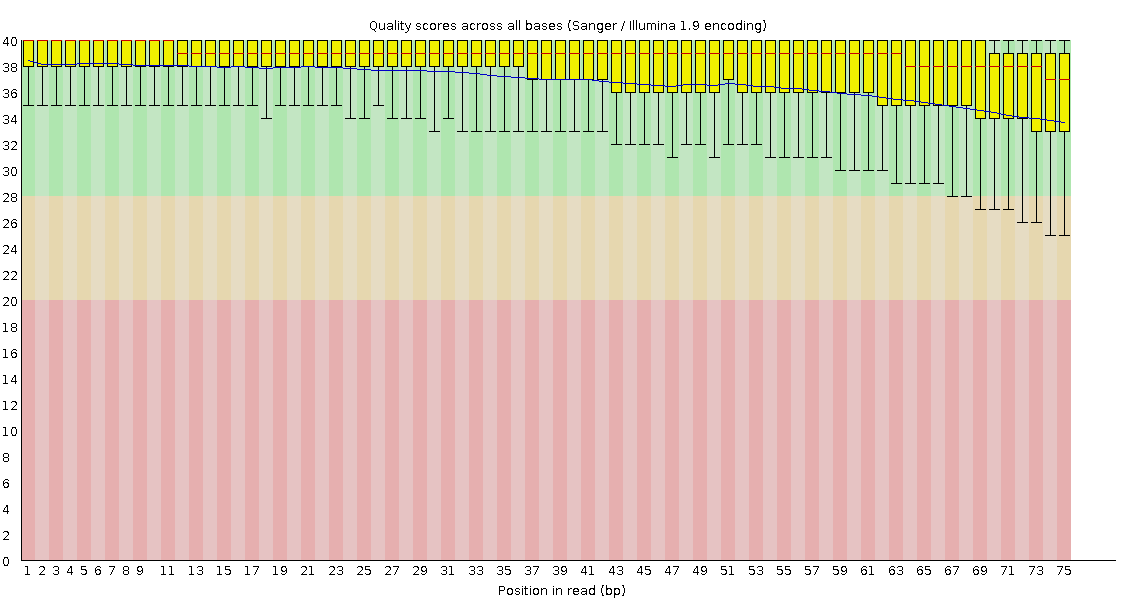
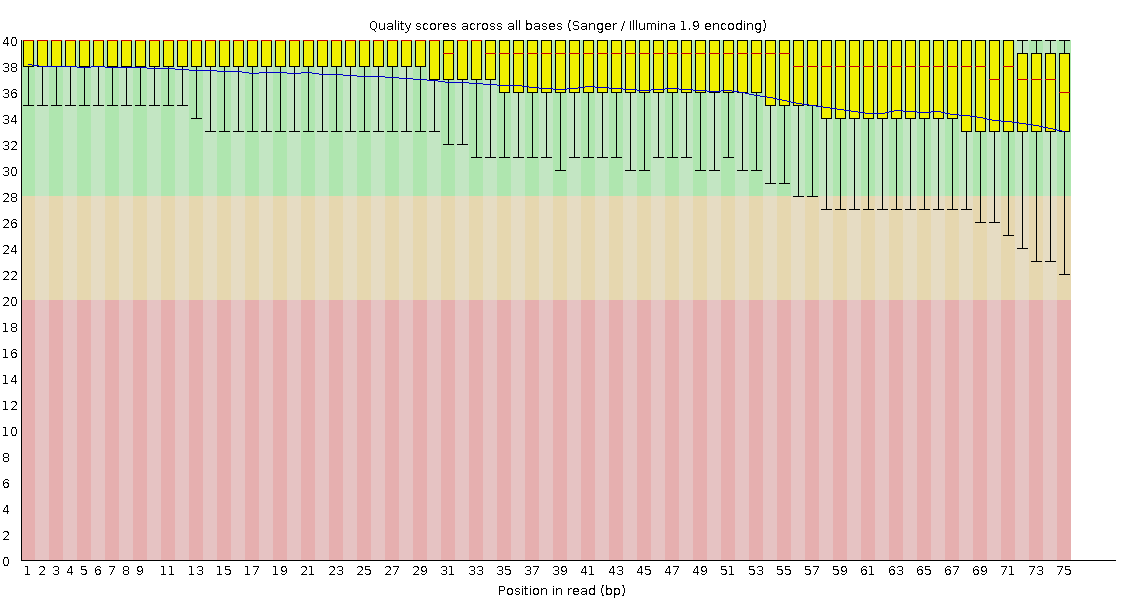
В обоих направлениях довольно хорошая, но обратные чуть похуже. По большей части всё (включая хвосты) в зелёной зоне, только в конце (с 67 позиции в прямом, с 57 в обратном) хвосты вылезают в жёлтый.
Per sequence quality scores
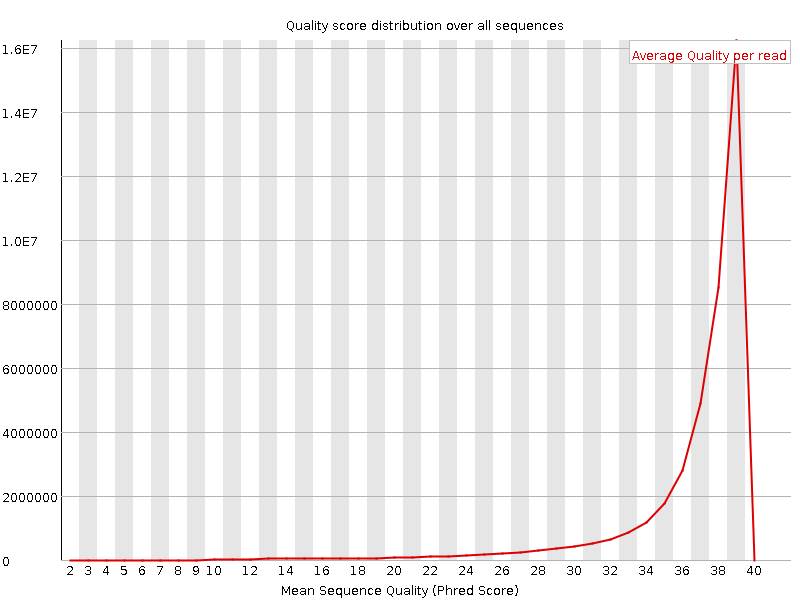
Тоже хорошая, чуть получше у прямых чтений. Там число ридов с наилучшим качеством (39 у обоих) повыше, кажется (по крайней мере отмеченно на графике максимальное деление 1.6Е7 против 1.4Е7, хотя график непонятно, где кончается - мешается надпись). И у обратных есть небольшой всплеск совсем некачественных.
Sequence Length Distribution
Одинаковая у обоих: все чтения длины ровно 75 bp.
5. Фильтрация чтений
java -jar /usr/share/java/trimmomatic.jar PE -phred33
../SRR10720416_1.fastq.gz ../SRR10720416_2.fastq.gz
SRR10720416_1_paired.fq.gz SRR10720416_1_unpaired.fq.gz
SRR10720416_2_paired.fq.gz SRR10720416_2_unpaired.fq.gz
TRAILING:20 MINLEN:50 &> trimmomatic.log
Версия 0.38, log-файл.
Параметры:
- PE - значит у меня парные концы
- phred33 - что у нас score в формате phred33
- Trailing:20 - удаляет с конца чтений нуклеотиды с качеством ниже 20
- MINLEN:50 - оставляет только такие чтения, длина которых не ниже 50 нуклеотидов
Остальное - просто название входных и выходных файлов.
6. Проверка качества триммированных чтений
fastqc -o fastqc_after_trim --extract trimmomatic_filtration/SRR10720416_*
&> fastqc_after_trim/fastqc.log
Версия FastQC v0.11.8, log-файл.
| 1_paired | 1_unpaired | 2_paired | 2_unpaired |
|---|---|---|---|
| 38 977 379 (95%) | 1 035 400 (2.5%) | 38 977 379 (95%) | 542 650 (1.3%) |
Per base sequence quality
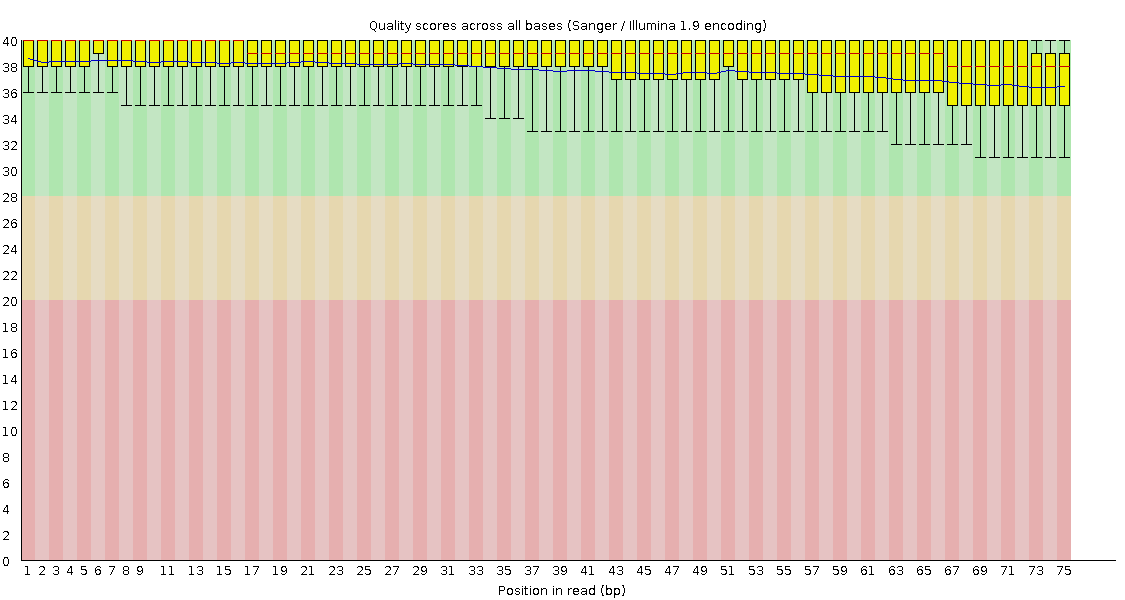
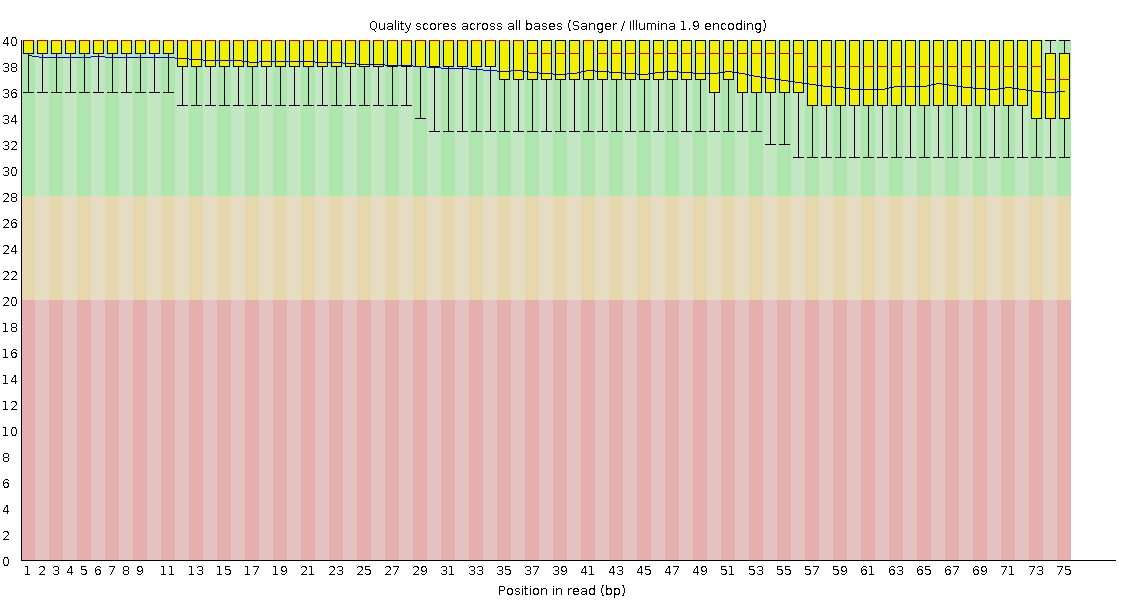

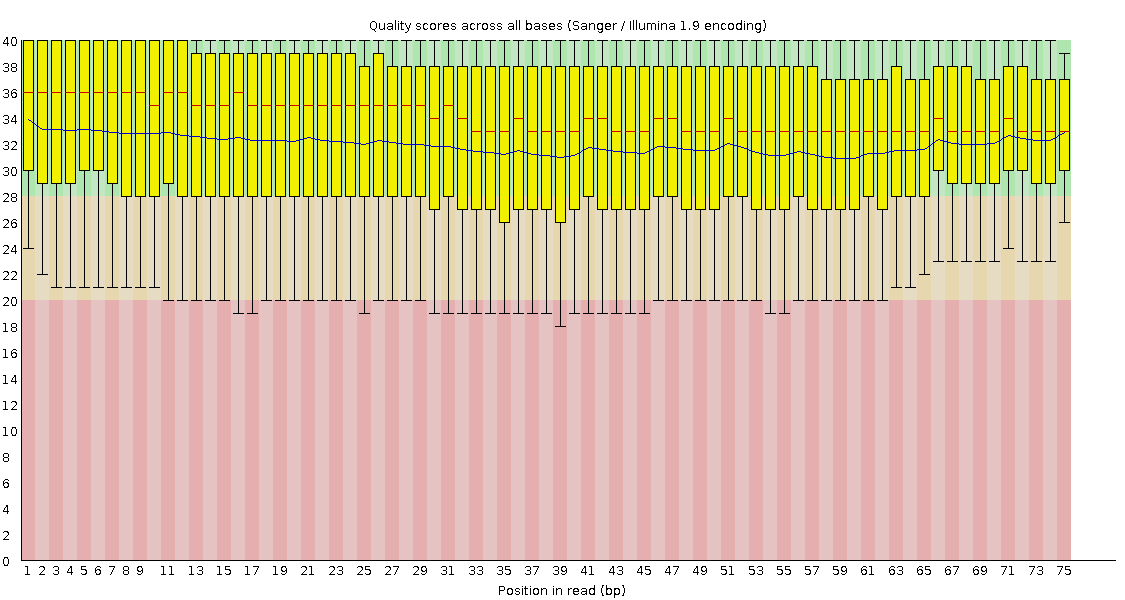
Paired vs. unpaired: unpaired заметно хуже - усы у paired целиком в зелёном, у unpaired практически все в жёлтом. И всё ещё у обртаных ещё хуже: ящики вылезают в жёлтое, усы в красное. В paired наблюдается ухудшение к концу в обоих случаях, в прямых unpaired +- тоже, а в обртаных unpaired скорее куда-то к середине.
Paired vs. исходные: после триммирования стало лучше. Всё ещё некоторое ухудшение у концу, но теперь только в пределах зелёной зоны. Обратные уже не выглядят чем-то хуже прямых (видимо, он был хуже за счёт тех, которые остались в unpaired), даже получше, если смотреть, как долго среднее держится на максимуме.
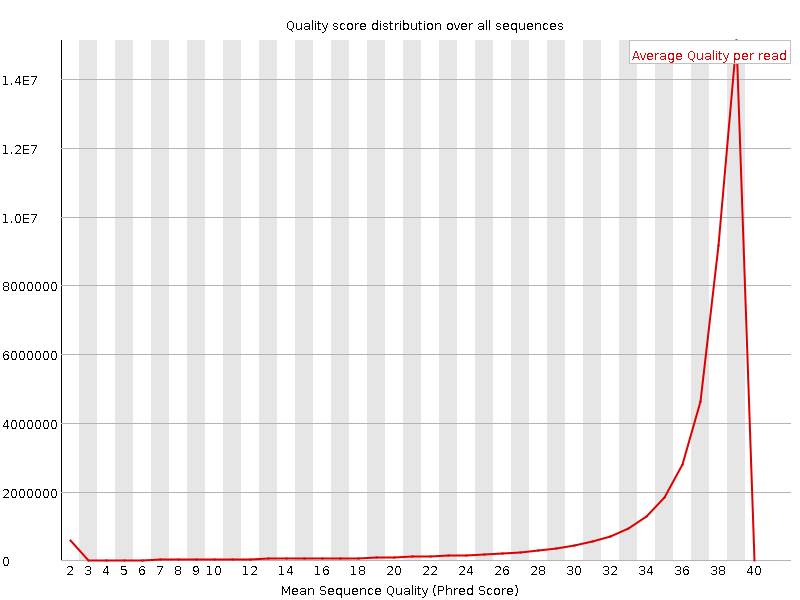
Per sequence quality scores
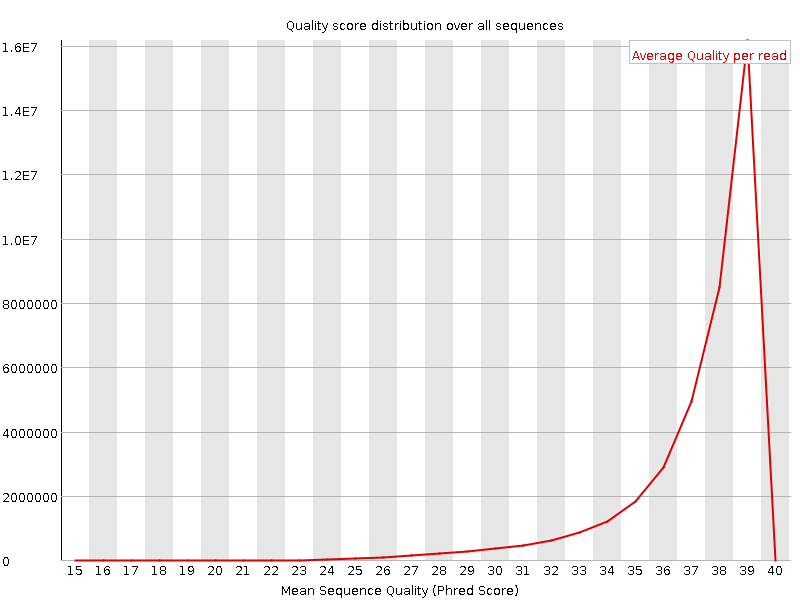
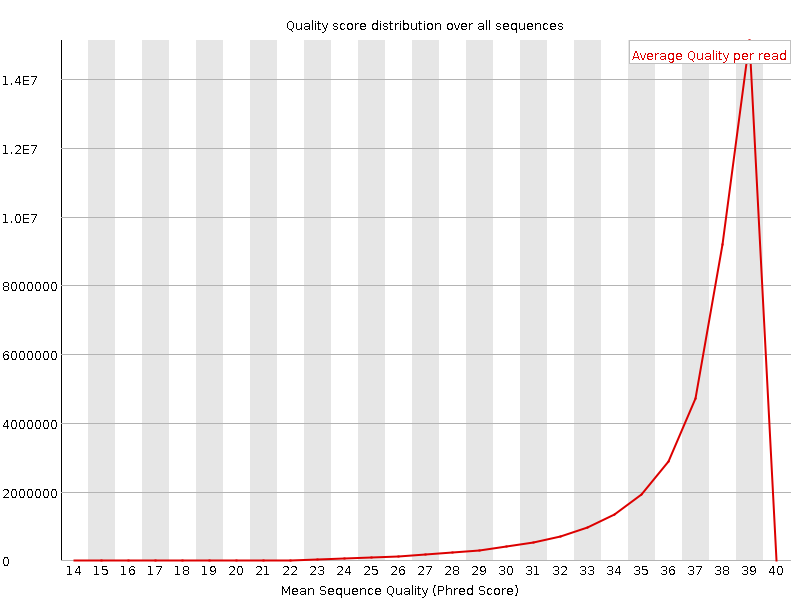
Стало лучше. Совсем плохих не осталось (график даже не рисует туда ось), начинается с 15 (прямые) или 14 (обратные). Ожидаемо исчез тот всплеск плохих у обратных. В остальном различий нет.
Sequence length distribution
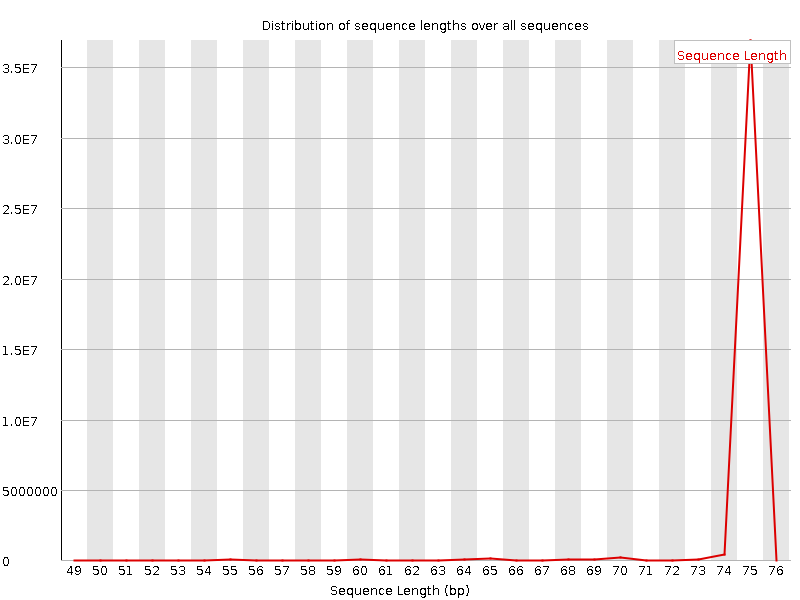
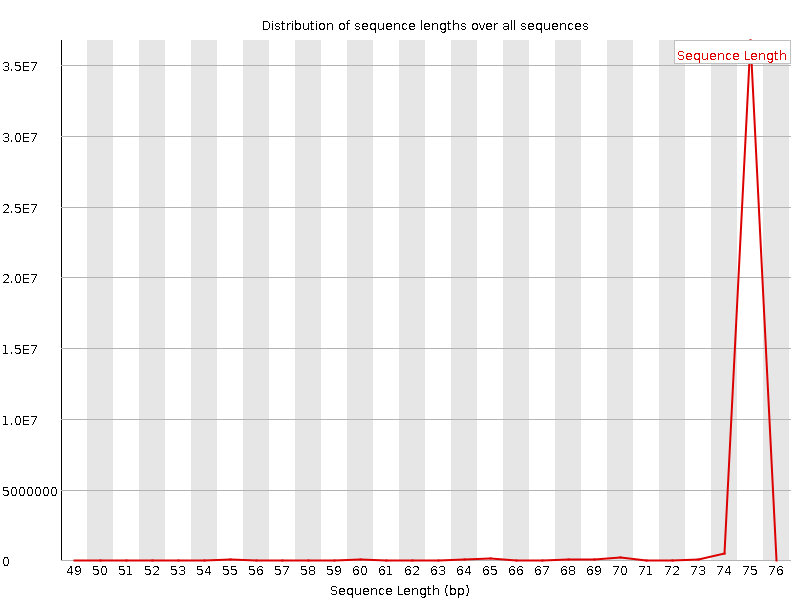
Картинка очень похожая в обоих случаях. Теперь (в результате обрезания плохих нуклеотидов) кроме чтений по 75 нуклеотидов появилось немного более коротких - вплоть до 49. Чуть заметные пики на 74, 70, 65.