Докинг низкомолекулярных лигандов в структуру белка
1
Задание было выполнено на примере лизоцима тюленя, структура которого была построена на основе гомологичного моделирования на прошлом практикуме (выбрана первая из построенных моделей).
В банке pdb была найдена SMILES нотация для NAG:
CC(=O)N[C@@H]1[C@H]([C@@H]([C@H](O[C@H]1O)CO)O)O
2
С помощью obgen была построена 3D структура лиганда в pdb формате:
obgen nag.smi > nag.mol babel -imol nag.mol -opdb nag.pdb
3..4
Скриптом prepare_ligand4.py были созданы pdbqt файлы лиганда и белка:
prepare_ligand4.py -l nag.pdb prepare_receptor4.py -r lys.pdb
5..6
Центр куба, в которм будет происходить поиск места для связывания, был определен с помощью создания псевдоатома. Таким образом был создан файл с параметрами докинга:
center_x=42.4 center_y=44.0 center_z=28.2 size_x = 25 size_y = 25 size_z = 25 num_modes = 20
И, собственно, запущен первый докинг:
vina --config vina.cfg --receptor prot.pdbqt \
--ligand nag.pdbqt --out nag_prot.pdbqt --log nag_lyz.log
7
Энергии трех лучших расположений и геометрическая разница между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.7 0.000 0.000
2 -5.6 2.106 4.691
3 -5.5 2.053 4.934Второе и третье расположение очень похожи между собой, но принципиально различаются с первым (что видно по rmsd).
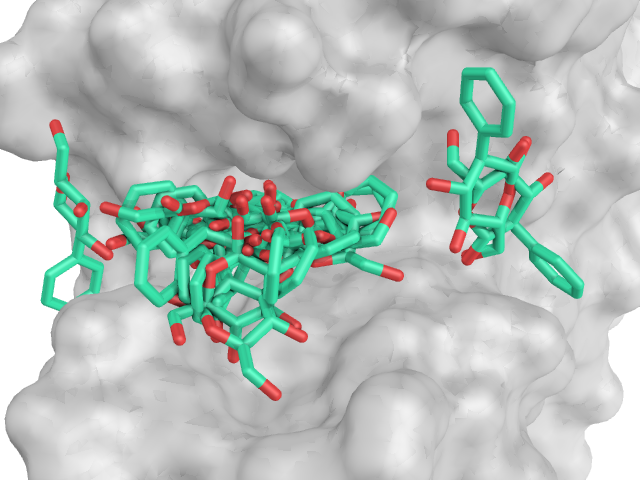
На картинке ниже отображены все состояния полученной структуры на одной картинке. Видно, что лиганду доступен определенный "карман", внутри которого его положение может меняться.
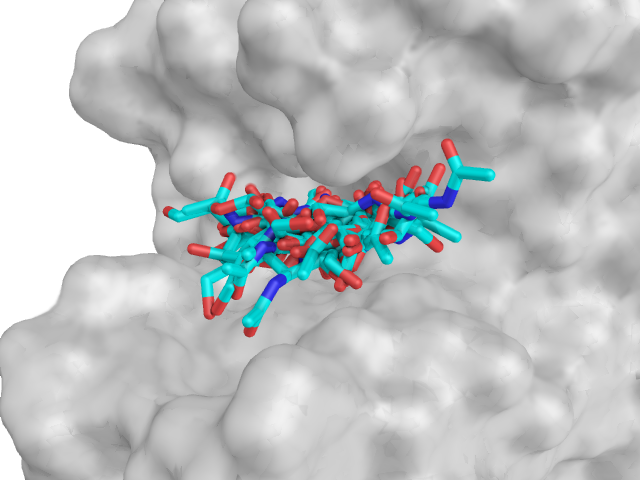
8
Затем был проведен докинг с учетом подвижности некоторых боковых радикалов белка, а именно трех аминокислот, по которым было проделано гомологичное моделирование в прошлом практикуме. Эти аминокислоты содержат атомы, образующие водородную связь с атомами лиганда.
python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py \
-r prot.pdbqt -s GLU53_ASN64_ASP120
vina --config vina.cfg --receptor prot_rigid.pdbqt --flex prot_flex.pdbqt \
--ligand nag.pdbqt --out nag_prot_flex.pdbqt --log nag_prot_flex.log9..10
Энергии трех лучших расположений и геометрическая разница между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.9 0.000 0.000
2 -5.8 1.283 2.486
3 -5.6 1.702 3.011
Энергия лучшего связывания лиганда с подвижным белком меньше, чем с полностью неподвижным (-5.9 против -5.7 ккал/моль).
В случае с подвижным белком лиганд входит в предоставленный ему "карман" менее глубоко, в 20-м представленном состоянии он даже не заходит в него. При этом первое положение лиганда в обоих случаях очень похоже; энергии отличаются, видимо, именно засчет движения аминокислот.
Докинг с подвижными остатками занял чуть больше времени, чем c полностью неподвижным белком.
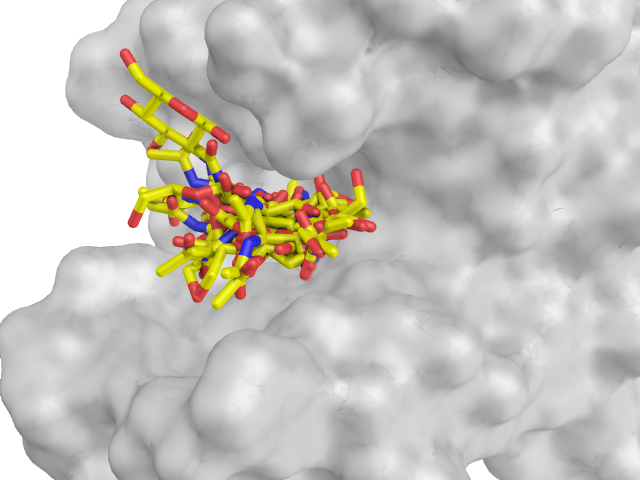
11
Аналогичные операции были проделаны еще для четырех лигандов, где СH3C(=O)NH группа была заменена на:
Скрипт (с выводом таблицы энергий и rmsd):
for i in {2..5};do
obgen nag${i}.smi > nag${i}.mol
babel -imol nag${i}.mol -opdb nag${i}.pdb
prepare_ligand4.py -l nag${i}.pdb
vina --config vina.cfg --receptor prot.pdbqt \
--ligand nag${i}.pdbqt --out nag${i}_prot.pdbqt --log nag${i}_prot.log
out=nag${i}_prot.log
echo -e "$out" >> tables.txt
grep 'mode' $out -A 5 >> tables.txt
done
Результат:
nag2_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.2 0.000 0.000
2 -4.7 1.596 3.897
3 -4.5 1.073 3.348
nag3_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.2 0.000 0.000
2 -5.2 2.127 3.791
3 -5.1 1.995 3.632
nag4_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.0 0.000 0.000
2 -4.9 2.453 3.202
3 -4.9 2.636 4.393
nag5_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.3 0.000 0.000
2 -6.2 2.067 3.067
3 -5.9 2.491 5.508
Если судить по энергиям наилучших состояний, лучше всего (и с большим отрывом) с белком связывается Ph-лиганд, хуже всего - H-лиганд. Все лиганды расположены в "кармане" белка одинаково глубоко. У OH-лиганда есть два основных расположения в пространстве (видно на картинке ниже), у остальных лигандов такого четкого разделения нет и они распределяются по всему предоставленному "карману".
OH (nag2) | NH2 (nag3) |
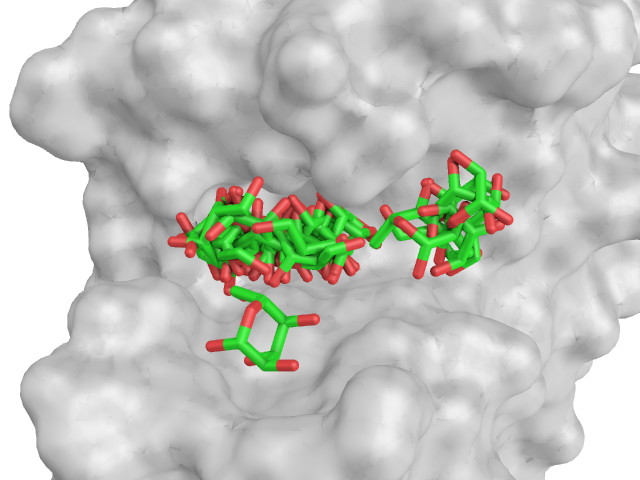 |
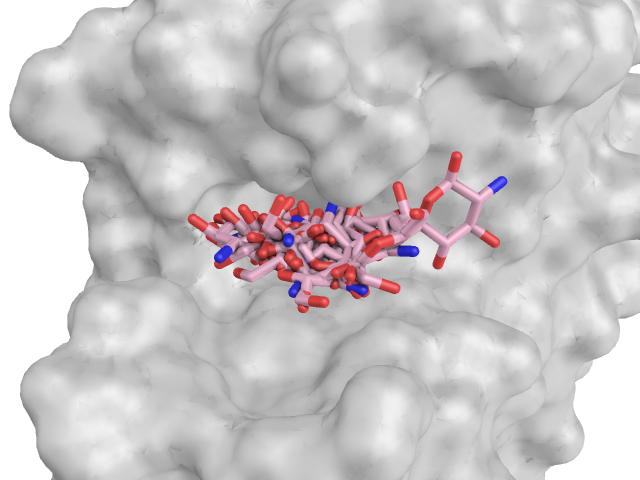 |
H (nag4) | Ph (nag5) |
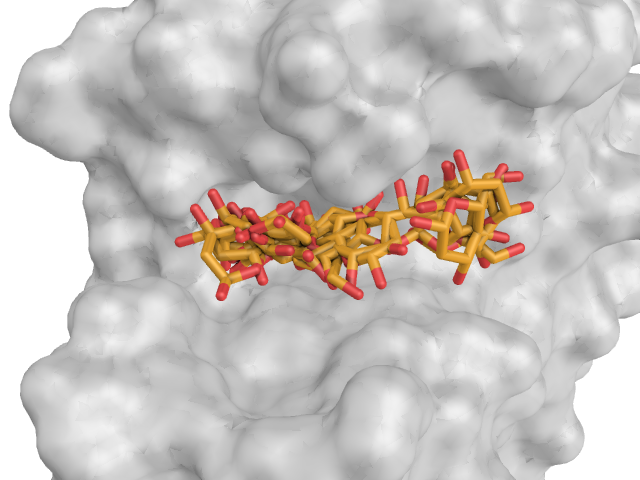 |
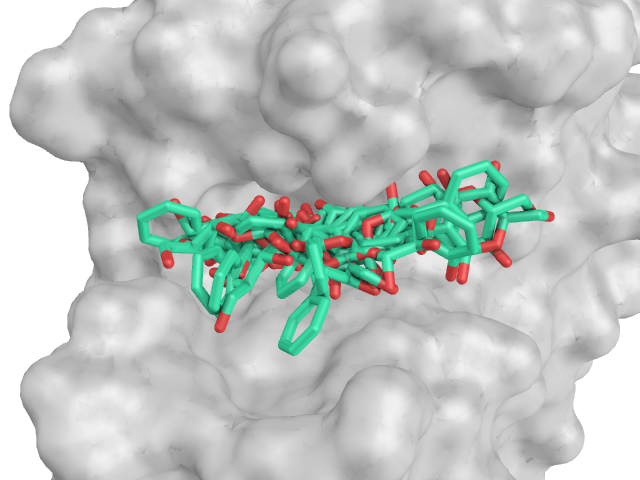 |
12
Был проведен докинг для OH-, NH2- и Ph-лиганда c подвижными радикалами белка.
nag2_prot_flex.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.2 0.000 0.000
2 -4.8 1.248 3.065
3 -4.6 1.135 2.766
nag3_prot_flex.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.3 0.000 0.000
2 -5.2 1.790 3.031
3 -5.1 1.627 3.029
nag5_prot_flex.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.5 0.000 0.000
2 -6.3 2.234 4.768
3 -6.1 2.964 5.277
Энергии лучших состояний понизились для всех лигандов. Изменилось и их расположение относительно белка. OH-лиганд часто вообще не входит в "карман" белка и не образует водородных связей с подвижными остатками; NH2-лиганд тоже поменял свое расположение относительно результатов с неподвижным белком; только Ph-лиганд остался более или менее там же. При этом различия наблюдаются в не-лучших состояниях, а расположение лиганда в состояниях с наименьшей энергией практически совпадает во свсех случаях. Энергия понижается по сравнению с белком без подвижных частей, видимо, как и в случае с полноценным nag, засчет движения подвижных остатков белка.
OH (nag2) | NH2 (nag3) |
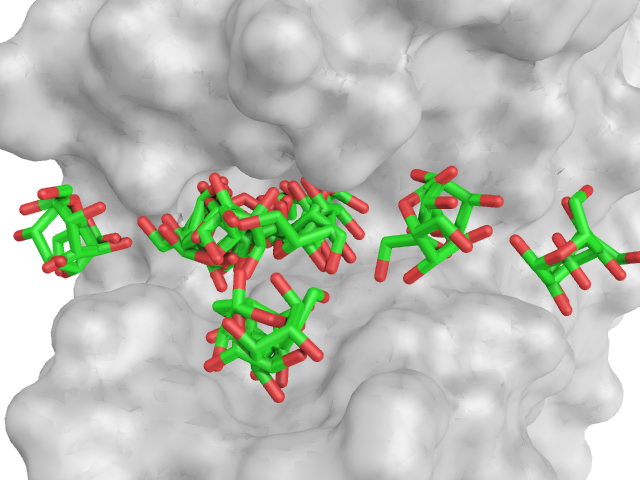 |
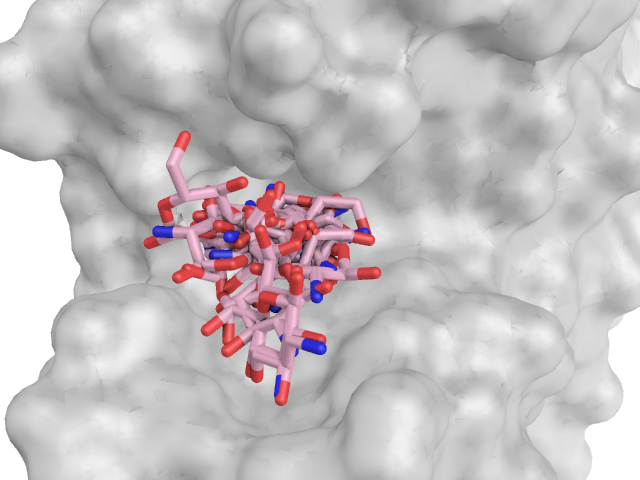 |
Ph (nag5) | |
 |