Молекулярная динамика биологических молекул в GROMACS
В качестве объекта моделирования было выбрано поведение ДНК в формамиде.
1..4
Файлы, использованные при моделировании:
- координаты дуплекса ДНК (А-форма), dna.pdb
- файл с ячейкой уравновешенных молекул формамида, fam_em.gro
- файл дополнительной топологии для формамида, fam.itp
- файл параметров для минимизации энергии, em.mdp
- файл параметров для "утряски" воды, pr.mdp
- файл параметров для молекулярной динамики, md.mdp
5
Вначале pdb файл со структурой ДНК-дуплекса был приведен к виду, который воспринимает GROMACS. Для этого название каждого нуклеотида было изменено (G → DG и так далее), атом C5M тимина переименован в C7, удалены фосфаты на 5'-концах. По получившемуся файлу dna.pdb была построена топология в силовом поле amber99sb и файл с координатами в формате GROMACS. Используемая модель воды - tip3p (из трех частиц).
pdb2gmx -f dna.pdb -o dna -p dna -ffamber99sb -water tip3p
6
С помощью editconf в ячейке был создан отступ от ДНК в 1.5 nm:
editconf -f dna.gro -o dna_ec -d 1.5
7
После этого была оптимизирована геометрия системы, чтобы удалить "плохие" контакты в молекуле:
grompp -f em -c dna_ec -p dna -o dna_em -maxwarn 1 mdrun -deffnm dna_em -v
Значение максимальной силы уменьшилось в ходе оптимизации геометрии:
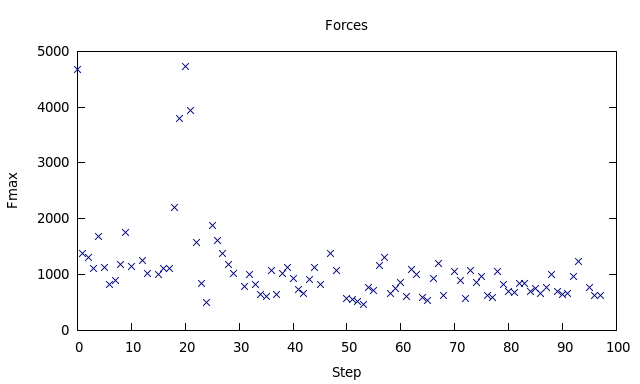
Step 0 Fmax = 4.684e+03 Step 1 Fmax = 1.375e+03 ... Step 97 Fmax = 6.318e+02
8
Добавлены молекулы формамида:
genbox -cp dna_em -p dna -cs fam_em.gro -o dna_s
Всего было добавлено 1289 молекул растворителя.
9
После этого файл топологии dna.top был вручную изменен: добавлена ссылка на itp формамида и число молекул формамида.
10
Для нейтрализации заряда системы был построен tpr (из вывода grompp был получен заряд системы = -10), а затем добавлено, соответственно, 10 положительно заряженных ионов (Na+).
grompp -f em -p dna -c dna_s -o dna_s genion -s dna_s -o dna_si -p dna -np 10
11
"Утряска" воды:
grompp -f pr -c dna_si -p dna -o dna_pr -maxwarn 1 mdrun -deffnm dna_pr -v
12
Файлы dna_si.gro (до утряски растворителя) и dna_pr.gro (после утряски) были переформатированы в pdb с помощью editconf. До утряски молекулы формамида располагались упорядоченно:

После утряски они расположены более или менее хаотично:

13..15
Был создан окончательный tpr файл для моделирования:
grompp -f md -c dna_pr -p dna -o dna_md -maxwarn 1
И, наконец, запущено само моделирование (в конечном итоге количество шагов составило 2000000, что соответствует 40 нс).