| Главная страница |
Анализ качества чтений
С помощью программы FastQC был проведен контроль качества чтений генома из 20 хромосомы.Команда fastqc chr20.fastq запускает работу программы и в качестве результата выдает различную информацию о чтениях:
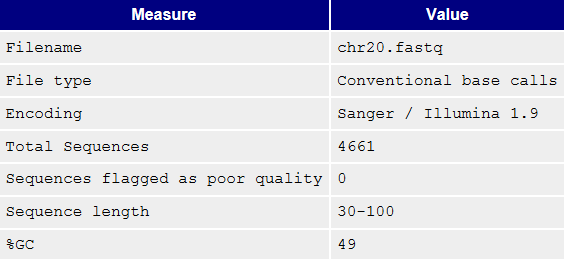
Графическое изображение качества определения нуклеотидов по позициям:
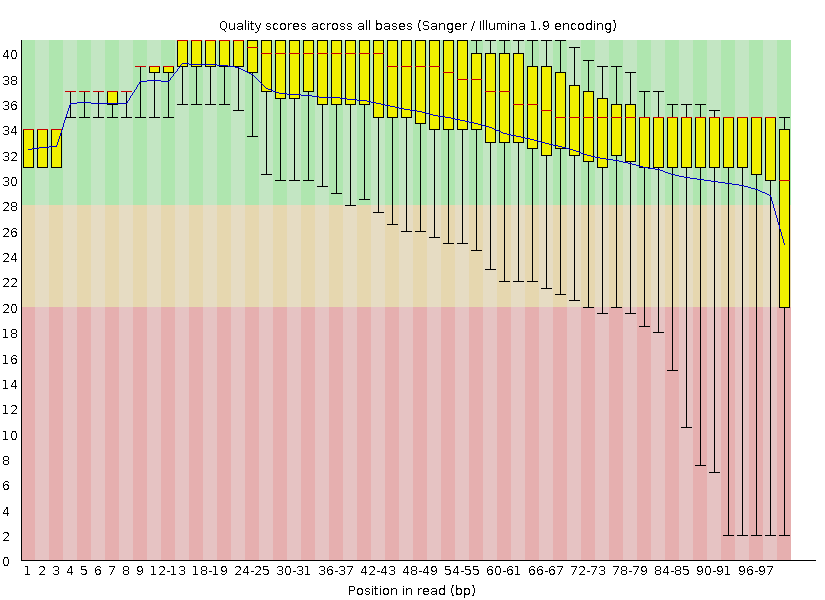
Видно, что с концов качество хуже.
Очистка чтений
С помощью программы Trimmomatic чтение было очищено: с конца удалены нуклеотиды с качеством ниже 20, и все чтения длиной меньше 50 нуклеотидов.Программа вызывалась командой java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr20.fastq chr20_tr.fastq TRAILING:20 MINLEN:50
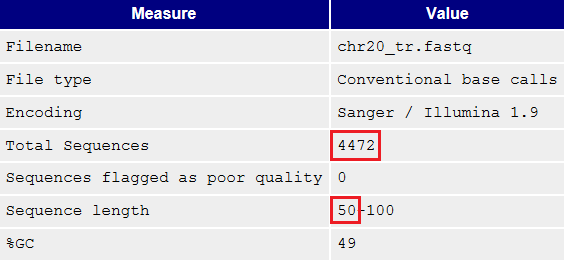
Удалено 189 чтений.
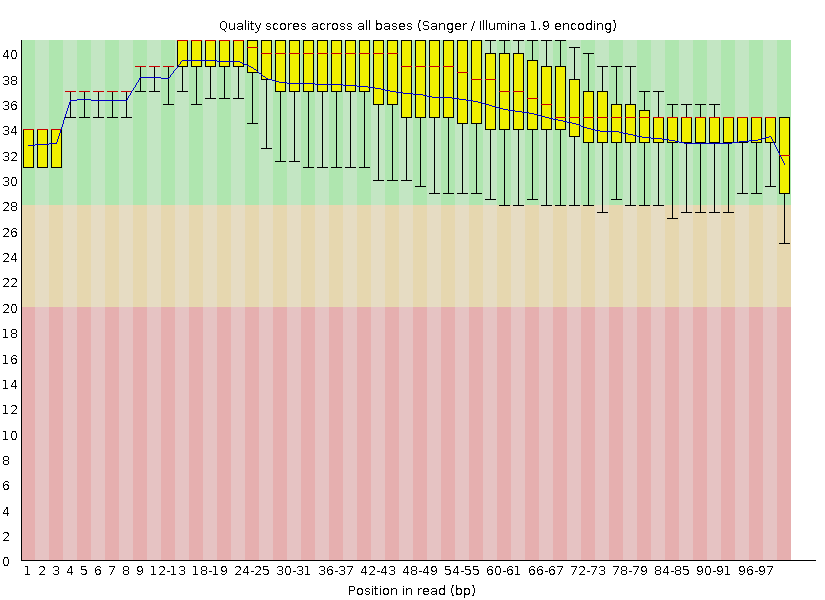
График вылезает из зеленой области в немногих местах и только "усами".
Картирование чтений с помощью программы BWA
С помощью команды bwa index chr20.fasta референсная последовательность была проиндексирована.Затем построено выравнивание прочтений и референса, команда: bwa mem chr20.fasta chr20_tr.fastq > chr20.sam.
Анализ выравнивания
Файл chr20.bam был переведен с бинарный форматом .bam с помощью команды samtools view -b -o chr20.bam chr20.sam.
Полученный файл был отсортирован по координате в референсе: samtools sort -T /tmp/chr20_sorted -o chr20_sorted.bam chr20.bam
Далее файл был проиндексирован: samtools index chr20_sorted.bam.
С помощью команды samtools idxstats chr20_sorted.bam было посчитано количество откартированных чтений.

На референсную 20 хромосому откартировалось 4468 чтений (не откартировались 4).
Поиск SNP и инделей
Файл с полиморфизмами был создан с помощью команды samtools mpileup -uf chr20.fasta chr20_sorted.bam -o chr20_snp.bcf.Командой bcftools call -cv chr20_snp.bcf -o chr20_snp.vcf создан файл со списком отличий между референсом и чтениями.
Всего 41 полиморфизм, все из них являются заменами.
Три из них:
| Координата | Тип полиморфизма | Референс | Чтения | Глубина покрытия | Качество чтений |
| 33961867 | замена | T | C | 19 | 150.008 |
| 33963485 | замена | G | C | 1 | 9.52546 |
| 33974207 | замена | A | G | 39 | 207.009 |
Аннотация SNP
Для работы с программой annovar из .vcf файла необходимо получить файл, с которым умеет работать эта программа. Это было сделано с помощью скрипта convert2annovar.pl, команда: ./convert2annovar.pl -format vcf4 chr20_snp.vcf -outfile 1_chr20.avinputДля аннотации файла с snp с помощью баз данных был использован скрипт annotate_variation.pl.
База данных refgene
Аннотация получена с помощью команды ./annotate_variation.pl -out 0_refgene -build hg19 1_chr20.avinput /nfs/srv/databases/annovar/humandb/
Все snp разделены на попадающие в экзоны (их 8), попадающие в интроны (32) и попадающие в 5'-нетранслируемую область (1). Вообще бывают следующие категории: exonic, splicing, ncRNA, UTR5, UTR3, intronic, upstream, downstream, intergenic. Также все sn p делятся на hom и het - их 23 и 18 соответственно. Для экзонных snp выделяют гомологичные и негомологичные замены. В данном случае их по 4.
Несинонимичные замены: в положениии 34022387 серин в 276 положении заменен на аланин в 56186884 - глутамин (183) на аргинин, в 56189985 - аспарагиновая кислота (79) на гистидин, в 56190634 - глутаминовая кислота (13) на лизин.
snp попали в следующие гены: GDF5 (growth differentiation factor 5), SPATA2 (spermatogenesis associated 2), ZBP1 (Z-DNA binding p rotein 1), UQCC1 (ubiquinol-cytochrome c reductase complex assembly factor 1).
Покрытие (и качество, соответственно, тоже) более чем у половины (у 24) snp плохое: 1-3, 9 замен имеют покрытие больше 20.
База данных dbsnp
Аннотация получена с помощью команды ./annotate_variation.pl -filter -out 0_dbsn p -build hg19 -dbtype snp138 1_chr20.avinput /nfs/srv/databases/annovar/humandb/
Имеют rs 30, не имеют 11.
База данных 1000 genomes
Аннотация получена с помощью команды ./annotate_variation.pl -filter -out 0_1000genomes -build hg19 -dbtype 1000g2014oct_all 1_chr20.avinput /nfs/srv/databases/annovar/humandb/
Аннотированных 30, неаннотированных 11 (те же самые, что и в dbsnp).
Частота встречаемости разная: от 1,4% до 99,6%.
База данных GWAS
Аннотация получена с помощью команды ./annotate_variation.pl -regionanno -out 0_gwas -build hg19 -dbtype gwasCatalog 1_chr20.avinput /nfs/srv/databases/annovar/humandb/
Три замены ассоциированы со следующими призраками человека: вес, псориаз, атрофия гиппокампа.
База данных Clinvar.
Аннотация получена с помощью команды ./annotate_variation.pl -filter -out 0_clinvar -build hg19 -dbtype clinvar_20140211 1_chr20.avin put /nfs/srv/databases/annovar/humand/
В этой базе данных ничего не нашлось.
Тaблица.
Использованные команды:
| fastqc chr20.fastq | анализ чтений |
| java -jar /usr/share/java/trimmomatic.jar SE | phred33 chr20.fastq chr20_tr.fastq TRAILING:20 MINLEN:50 – очистка чтений: с конца удалены нуклеотиды с качеством ниже 20, и все чтения длиной меньше 50 нуклеотидов |
| bwa index chr20.fasta | индексирование референсной последовательности |
| bwa mem chr20.fasta chr20_tr.fastq > chr20.sam | построение выравнивания прочтения и референса |
| samtools view -b -o chr20.bam chr20.sam | перевод файла в бинарный формат .bam |
| samtools sort -T /tmp/chr20_sorted -o chr20_sorted.bam chr20.bam | сортировка по координате |
| samtools index chr20_sorted.bam | индексирование |
| samtools idxstats chr20_sorted.bam | подсчет количества откартированных чтений |
| samtools mpileup -uf chr20.fasta chr20_sorted.bam -o chr20_snp.bcf | создание файла с полиморфизмами |
| bcftools call -cv chr20_snp.bcf -o chr20_snp.vcf | создание файла со списком отличий между референсом и чтениями |
| ./convert2annovar.pl -format vcf4 chr20_snp.vcf -outfile 1_chr20.avinput | создание файла, с которым может работать annovar |
| ./annotate_variation.pl -out 0_refgene -build hg19 1_chr20.avinput /nfs/srv/databases/annovar/humandb/ | аннотирует файл по базе |
| ./annotate_variation.pl -filter -out 0_dbsnp -build hg19 -dbtype snp138 1_chr20.avinput /nfs/srv/databases/annovar/humandb/ | аннотирует по базе данных dbsnp |
| ./annotate_variation.pl -filter -out 0_1000genomes -build hg19 -dbtype 1000g2014oct_all 1_chr20.avinput /nfs/srv/databases/annovar/humandb/ | аннотирует файл по базе данных 1000genomes |
| ./annotate_variation.pl -regionanno -out 0_gwas -build hg19 -dbtype gwasCatalog 1_chr20.avinput /nfs/srv/databases/annovar/humandb/ | аннотирует файл по базе данных GWAS |
| ./annotate_variation.pl -filter -out 0_clinvar -build hg19 -dbtype clinvar_20140211 1_chr20.avinput /nfs/srv/databases/annovar/humand | аннотирует файл по базе данных Clinvar |