Практикум 3: хемоинформатика¶
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit import RDConfig
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
import numpy as np
from IPython.display import display,Image
import rdkit.Chem.Lipinski as Lipinksy
# Does molecule suits Lipinsky rule?
def True_Lipinsky(mol):
don = Lipinksy.NumHDonors(mol) <= 5
acc = Lipinksy.NumHAcceptors(mol) <= 10
mw = Lipinksy.rdMolDescriptors.CalcExactMolWt(mol) <= 500
coef = Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(mol)[0] < 5
if don and acc and mw and coef:
return True
Построим ибупрофен по SMILES
ibu_form = 'CC(C)CC1=CC=C(C=C1)C(C)C(=O)O'
ibu=Chem.MolFromSmiles(ibu_form)
AllChem.Compute2DCoords(ibu)
display(ibu)

Проверим, что он соотвествует правилам Липински:
- Молекула имеет не более 5 доноров водородных связей
- Молекула имеет не более 10 акцепторов водородных связей
- Молекулярная масса менее 500 Да
- log P (мера липофильности, коэффициент распределения в системе октанол-1 - вода) меньше 5.
print(Lipinksy.NumHDonors(ibu))
print Lipinksy.NumHAcceptors(ibu)
print Lipinksy.rdMolDescriptors.CalcExactMolWt(ibu)
print Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(ibu)[0]
Теперь нарисуем производное ибупрофена, которое могло бы участвовать в азид-алкиновом циклоприсоединении. Для этого в молекуле должна быть концевая тройная связь.
ibu_m_f = 'C#CCC1=CC=C(C=C1)C(C)C(=O)O'
ibu_m=Chem.MolFromSmiles(ibu_m_f)
AllChem.Compute2DCoords(ibu_m)
display(ibu_m)

Теперь сделаем фрагмент, кторый будет получаться после реакции циклоприсоединения.
ibu_m_f = 'N1C=C(CC2=CC=C(C=C2)C(C)C(=O)O)N=N1'
ibu_m=Chem.MolFromSmiles(ibu_m_f)
AllChem.Compute2DCoords(ibu_m)
display(ibu_m)

import pubchempy as pcp
Теперь найдем радикалы с азидом, подходящие для клик-химии. Я попробовала скачать необходимые вещества программно, но не разобралась, как задавать дополнительные опции поиска. Поэтому здесь привожу не совсем удачный вариант кода, сам файл со SMILES'aми получила скачала вручную.
azide = '[N-]=[N+]=[N-]'
per_page = 10**2
list = []
for i in range(20):
try:
list1 = pcp.get_compounds(azide,
'smiles',
searchtype='substructure',
listkey_count = per_page,
listkey_start = i*per_page
)
except:
pass
list.extend(list1)
print(len(list))
Выделим из полученного файла SMILES'ы без разрывов, не слишком динные (и избавимся от дефектных)
strings=np.genfromtxt('pr3/smiles.txt',dtype=np.str)
smiles = []
for line in strings:
if len(line[1]) < 30 and not '.' in line[1] and len(line[1])>5:
smiles.append(line[1])
print(smiles[:10])
print(len(smiles))
Построим новые молекулы, используя полученные радикалы и измененный ибупрофен. Сохраним в отдельный список те, которые удовлетворяют правилам Липински.
azide = 'N=[N+]=[N-]'
right_mol = []
right_mol_Lipinsky = []
for smi in smiles[:1500]:
if azide in smi:
newsmi=smi.replace('N=[N+]=[N-]',ibu_m_f)
right_mol.append(newsmi)
else:
continue
try:
newmol=Chem.MolFromSmiles(newsmi)
if True_Lipinsky(newmol):
right_mol_Lipinsky.append(newmol)
except:
pass
Нарисуем 50 первых молекул, проверим, что все в порядке.
print(len(right_mol_Lipinsky))
Draw.MolsToGridImage(right_mol_Lipinsky[:50], molsPerRow=5)

Теперь построим карту схожести ибупрофена с веществами из массива.
from rdkit.Chem.Draw import SimilarityMaps
fig, maxweight = SimilarityMaps.GetSimilarityMapForFingerprint(ibu, right_mol_Lipinsky[5], SimilarityMaps.GetMorganFingerprint)
print(maxweight)

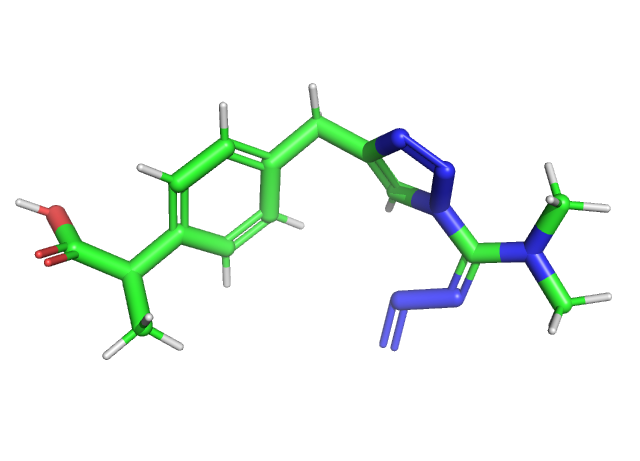
Построим 3D-структуру данной молекулы
m3d=Chem.AddHs(right_mol_Lipinsky[5])
Chem.AllChem.EmbedMolecule(m3d)
AllChem.MMFFOptimizeMolecule(m3d,maxIters=500,nonBondedThresh=200 )
Попробовала посмотреть в nglview, но почему-то не получилось.
import nglview as nv
nv.show_rdkit(m3d)
Поэтому воспользуемся паймолом. Файл я получила с помощью программы obgen из пакета openbabel:
obgen mol_3d.smi > mol_3d_ready.mol
f = open('mol_3d.smi', 'w')
f.write(Chem.MolToSmiles(m3d))
f.close()
from xmlrpclib import ServerProxy
import __main__
__main__.pymol_argv = [ 'pymol', '-x' ]
import pymol
pymol.finish_launching()
from pymol import cmd,stored
cmd.load('mol_3d_ready.mol')
cmd.orient()
cmd.bg_color('white')
cmd.png(filename='1', ray=1)