Анализ транскриптомов. Bedtools.
В ходе выполнения практикума было использовано множество команд, которые можно увидеть в таблице 1.
| Команда | Для чего была использована |
|---|---|
| fastqc chr2.1.fastq, fastqc chr2.1_2.fastq | анализ качества чтения, дает информацию о количестве ридов и их качестве |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr2.1.fastq chr2.1_2.fastq TRAILING:20 MINLEN:50 | были убраны все нуклеотиды с конца, имеющие качество прочтения ниже 20 и все риды, длина которых меньше 50 |
| hisat2 -x chr2 -U chr2_2.fastq -S chr2_alignment.sam --no-softclip | построение выравнивания референсной последовтельности и прочтения в формате sam. Параметр --no-spliced-alignment был убран, так как мы работаем с последовательностью РНК-траскриптов, из которых при созревании вырезаются интроны, поэтому они могут картироваться на геном с разрывами.-X - индексированная референсная последовательность, после -U указываем на файл с прочтениями, после S - название выходного файла. |
| samtools view -b chr2_alignment.sam -o chr2_alignment.bam | перевод файла с выравниванием в бинарный файл с форматом .bam. -o - название выходного файла, -b - перевод файла в бинарный формат. |
| samtools sort chr2_alignment.bam chr2_sorted | сортировка выравнивания чтений с референсом по координате начала чтения в референсе. Выходной файл - chr2_sorted.bam |
| samtools index chr2_sorted.bam | индексирование сортированного предыдущей командой файла chr2_sorted.bam.bai |
| htseq-count -f bam -s no -i gene_id -m union chr2_sorted.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseqc_results | Команда для подсчета чтений. Опция -f - формат файла с выравниванием (bam, sam). По умолчанию стоит sam. Опция -s указывает на цепь, по которой были выравнены риды. Из трех возможных вариантов (yes, reverse, no) был выбран вариант no, так как нам точно неизвестно, по какой из цепей строилось выравнивание. -i - GFF атрибут, используемый как feature-ID (нам подходит вариант по умолчанию). Последняя опция -m определяет режим работы команды для неоднозначных выравниваний. |
| grep -w 0 -v htseqc_results > allres.txt | Опция -w 0, чтобы выбрать строки с нулем, ограниченным пробелами, и -v, чтобы вывести все остальные. |
Задание 1. Анализ качества чтений и их очистка.
Был выбран файл chr2.1.fastq. Он содержал 12507 ридов, длина которых составляла от 28 до 51 нуклеотидов (хотя большинство содержали именно 51 нуклеотид), а среднее качество прочтения - около 39%. После очистки чтений их количество практически не уменьшилось: вырезалось 108 ридов (0,86% от всех), а длина стала лежать в диапазоне от 50 до 51 нуклеотида. На рисунках 1 и 2 можно видеть более информативную оченку качества чтений, полученную с помощью программы FastQC. По ним видно, что данное триммирование не было необходимым, так как графики практически не отличаются. Это же показывают и ранее упомянутые данные. Но в дальнейшем использовался файл с очищенными прочтениями.
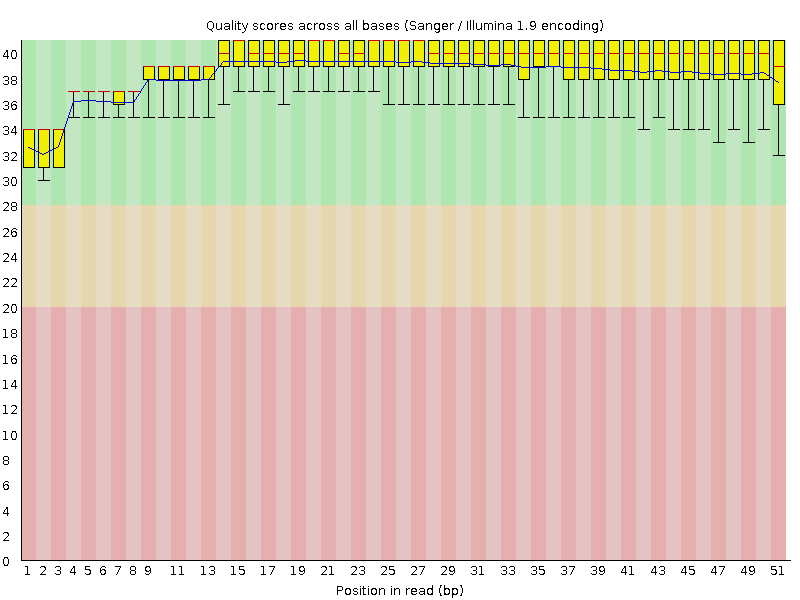
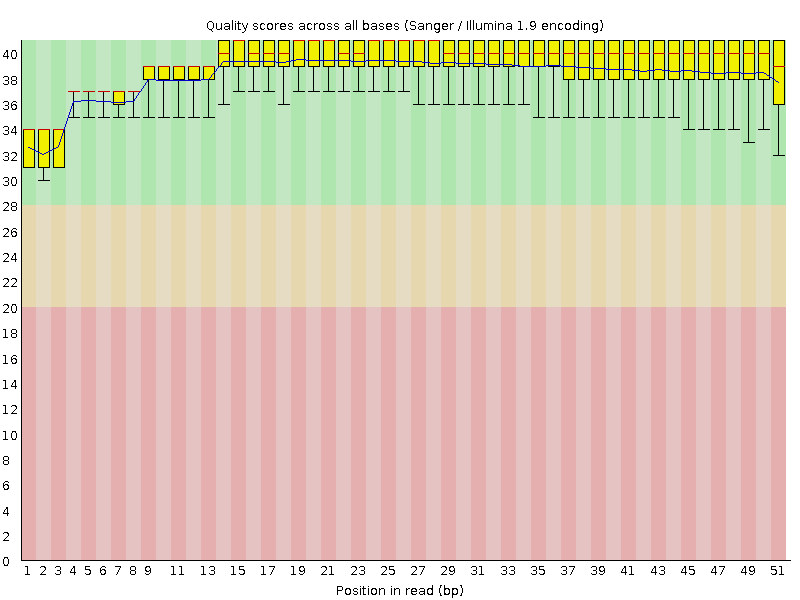
Задание 2. Картирование ридов.
Индексные файлы были просто скопированы в директорию, так как в этом практикуме работа идет с той же хромосомой. Потом было произведено картирование чтений. По выводу программы Hisat2:
12399 (100.00%) were unpaired; of these:
54 (0.44%) aligned 0 times
12345 (99.56%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.56% overall alignment rate
видно, что 99,56% чтений выравнились ровно один раз, всего 54 рида не выравнились ни разу. Можно сказать, что качество картирования высокое.
Задание 3. Анализ результатов.
Программа htseq-count выдает файл со строками, в которых указаны названия генов и количество прочтений, которые легли на этот ген. Чтобы проанализировать этот результат была использована команда grep, которая записала в файл allres.txt только строчки, в которых не встречается 0. Как видно:
ENSG00000115053.11 12058 ENSG00000202400.1 5 ENSG00000206885.1 12 ENSG00000233538.1 2 __no_feature 268 __not_aligned 54большинство ридов легли на один ген. Есть 54 прочтения, которые на предыдущих шагах не картировались на хромосому ни разу, и 268 прочтений, для которых границы генов не определились. Ген ENSG00000115053.11 имеет 14 экзонов и кодирует фосфопротеин, который участвует в синтезе и созревании рибосом и расположен в ядрышке. Примечательно, что его 11 интрон кодирует небольшую ядрышковую РНК. Ген ENSG00000202400.1, на который легло пять прочтений, кодирует белок SNORD82 - малую ядрышковую РНК, как и ген ENSG00000206885.1 - продукт (SNORA75). Транскрипт последнего гена относится к некодирующим РНК.