Практикум 11. Алгоритмы выравнивания
Задание 1. Сравнение гомологичных и негомологичных белков
В этом практикуме я сравнивала алгоритмы выравнивания - локальный (water) и глобальный (needle) алгоритмы выравнивания, а также училась определять гомологичность белков по построенным выравниваниям. Для этого я взяла по 5 пар гомологичных и негомологичных белков из протеомов Bacillus subtilis 168 (BACSU) и Escherichia coli K12 (ECOLI), полученных из Swissprot и проанализировала результаты глобального выравнивания (needle) и локального (water) этих белков. Негомологичные белки я старалась подбирать максимально похожие по функциям и длине, потому что именно задача разделения похожих, но не гомологичных белков чаще всего стоит перед исследователями. Параметры обоих выравниваний стандартные: gap opening penalty 10, gap extension penalty 0.5, matrix EBLOSUM62. Примеры выравниваний белков MURG - рис. 1, рис. 2.
У гомологичных белков бактерий, как правило, больший процент идентичности и схожести, но в негомологичных белках также встречается высокий процент схожести, так что ориентироваться только по этим параметрам, думаю, не стоит. Значительно отличает гомологичные белки от негомологичных высокий score, который учитывает функциональную консервативность (в зависимости от длины белка точные значения разнятся, так как он вычисляется по мономерам), достаточно малое число гэпов (менее 15%) и практически полное покрытие (выравниваемый участок белка для water практически соответствует самому белку). При этом стоит заметить, что существуют концевые гэпы, которые не всегда имеют большое значение для функционала белка (water, например, их не учитывает, также он не учитывает плохо выравниваемые участки в начале и в конце), но позволяют точнее отделять гомологичные белки от негомологичных.
В качестве вывода стоит сказать, что для оценки гомологичности белков лучше использовать глобальное выравнивание по алгоритму needle, а для оценки сходства их функций (или функций отдельных участков - например, АТФ-азного домена) лучше брать локальное выравнивание по алгоритму water (для оценки сходства отдельных доменов также ограничив выравнивание участком белка, в котором он находится).


Задание 2. Множественное выравнивание и сравнение выравниваний, построенных разными программами
В этой части практикума я производила множественное выравнивание по алгоритму muscle и сравнение выравнивания двух белков, полученного из множественного, с выравниваниями по уже знакомым алгоритмам needle и water. Для выполнения задания 2 необходимо было сначала построить множественное выравнивание нескольких гомологичных белков из разных огранизмов. Я выбрала достаточно консервативный белок ATPA (альфа-субъединица АТФ-синтазы), присутствующий у бактерий и в митохондриях и пластидах эукариот, из стандартных организмов генетиков, а также акантамёбы (Escherichia coli K12, Acanthamoeba castellanii, Saccharomyces cerevisiae, Caenorhabditis elegans, Homo sapiens, Arabidopsis thaliana). На множественном выравнивании алгоритмом muscle со стандартными настройками белки совершенно разных организмов оказались весьма похожими (50% идентичности, 60% сходства, менее 15% гэпов), так что можно утверждать, что они действительно гомологичны, хотя и не полностью консервативны.
Поскольку у эукариот этот белок встречается только в митохондриях и хлоропластах, то в выравнивание я взяла их предшественники (precursor), что, скорее всего, и объясняет аминокислотный "хвост" у митохондриальных белков (Acanthamoeba castellanii, Saccharomyces cerevisiae, Caenorhabditis elegans, Homo sapiens). Белок из хлоропластов Arabidopsis thaliana представлен самим собой, не предшественником, и подобного "хвоста" у него нет.
Ссылка на скачивание проекта JalView с множественным выравниванием.
Кроме того, я построила дерево на основе выравнивания с помощью встроенного инструмента со стандартными настройками (матрица весов BLOSUM62), и оно получилось верным. Думаю, это хорошо иллюстрирует, как работают филогенетики - здесь достаточно далекие организмы можно разместить на дереве всего по одному белку, а если взять больше белков, то можно будет построить более подробную схему для всё более близкородственных организмов.
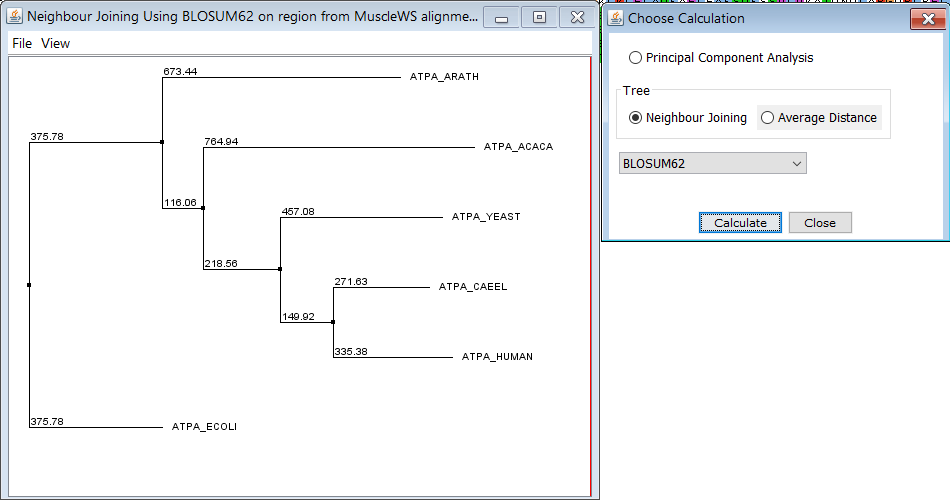
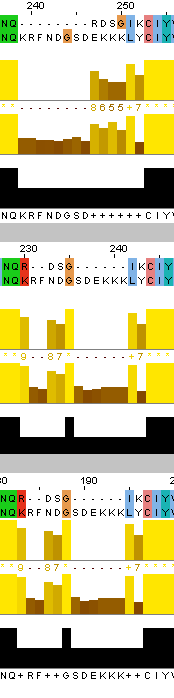
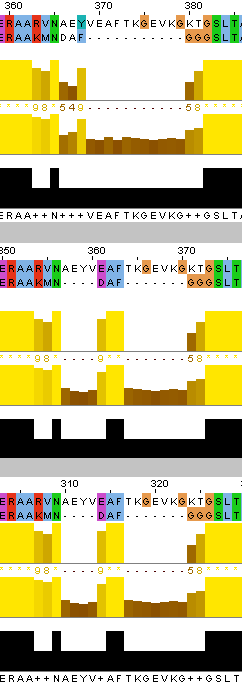
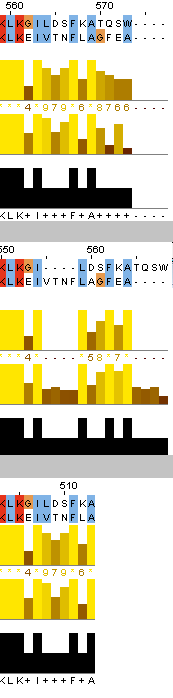
После этого я также выровняла наиболее далёкие друг от друга белки - ATPA_ECOLI и ATPA_HUMAN по алгоритмам needle и water, после чего сравнила полученные выранивания между собой и с выравниванием, полученным из множественного. Было найдено суммарно три отличия в расположении инделей, причём различия 1 и 2 - между множественным выравниванием и остальными, а различие 3 - между выравниваниями needle и water (множественное здесь совпало с water).
Отличие 1: muscle (множественное выравнивание) - индел у ECOLI на позициях 230-237 (сдвиг нумерации из-за другого белка в множественном выравнивании); needle и water - два индела 231-232 и 236-241 (сдвиг нумерации water). Отличие 2: muscle - индел у HUMAN на позициях 360-370; needle и water - два индела 357-360 и 364-370. Отличие 3: needle - индел 555-558 у ECOLI, индел 565-568 у HUMAN; mudcle и water - инделей нет. Стоит отметить, что, как и в задании 1, water не учитывает "лишние" аминокислоты в начале и конце пептида.
Ссылка на скачивание проекта JalView со сравнением выравниваний.
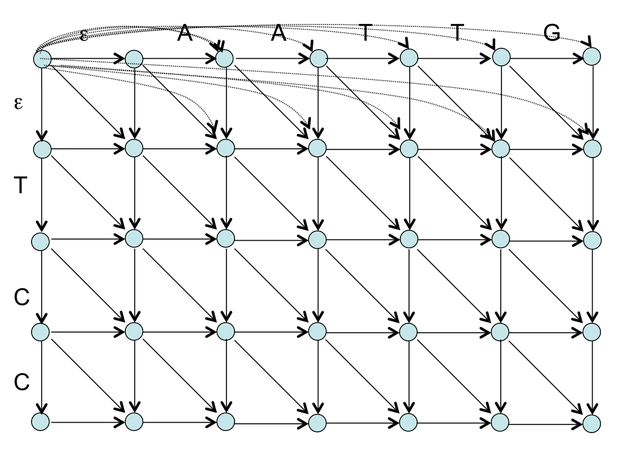
Задание 4. Описание ориентированного графа оптимального глобального выравнивания с аффинными штрафами за индели
Граф глобального выравнивания строится для всего белка, и все гэпы в начале/конце полностью учитываются. В начале гэпа граф переходит по оси z, и это даёт gap opening penalty, при этом каждый последующий гэп в инделе даёт только меньшее по модулю gap extension penalty, а возвращение из инделя на плоскость выравнивания (обратное движение по оси z) не даёт штрафа.
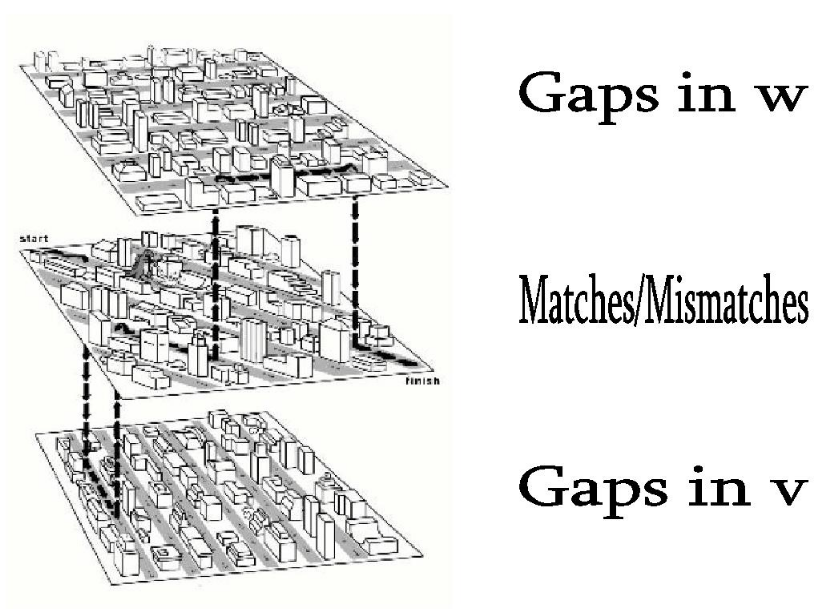
Задание 5. Описание ориентированного графа оптимального локального выравнивания с линейными штрафами за индели
В матрицу добавляются переходы с весом 0, ведущие из начальной точки во все клетки матрицы и из всех клеток в конечную точку (часто называемые в литературе free ride). Частичные графические интерпретации, как мне кажется, могут ввести в заблуждение (но одна всё же приведена), а полные слишком перегружены линиями. Художественное описание может сработать лучше: представьте, что вы едете по улицам Манхэттена посетить любимые магазины. В начале вы можете долететь на вертолёте до нужного квартала, а потом в любой момент опять вызвать вертолёт и долететь до конца маршрута. Магазинов (хорошего выравнивания) в воздухе нет, но и пробок (огромных инделей или плохо выравниваемых последовательностей в начале и конце) тоже. Линейные штрафы за гэпы описываются достаточно просто - все гэпы в инделе имеют равный вес (как будто каждый из них возник самостоятельно), соответственно, равный gap penalty.
Ссылки на источники
- http://www.csbio.unc.edu/mcmillan/Comp555S16/Lecture14.html – лекция-источник рисунка и графа для иллюстрирования (сайт Леонарда МакМиллана)