Практикум 12. BLAST
Задание 1. Нахождение гомологов в базе данных SwissProt с помощью BLAST
Мои параметры:
- Query length: 820
- Query subrange: N/A (default)
- Align two or more sequences: no (default)
- Algorithm: blastp (default)
- Organism: N/A (default)
- Exclude: N/A (default)
- Max target sequences: 20000 (default: 100)
- Short queries: no (defauld: yes)
- Expect treshhold: 10 (default)
- Word size: 6 (default)
- Max matches in a query range: 0 (default)
- Matrix: BLOSUM62 (default)
- Gap opening cost: 11 (default)
- Gap exstension cost: 1 (default)
- Compositional adjustments: conditional compositional score matrix adjustment (default)
- Genetic Code: 1 (default)
- Filter low complexity regions: no* (default)
- Mask for lookup table only: no (default)
- Mask lower case letters: no (default)
- Database: Non-redundant UniprotKB/Swiss-Prot(swissprot).
Дополнительная информация по параметрам (кроме очевидных):
Algorithm - используемый алгоритм BLAST (задание 3).
Organism - организм или таксономическая группа, белки которых мы рассматриваем (или наоборот, не рассматриваем).
Exclude - не учитывать в поиске некоторые группы белков. Задание 3.
Max target sequences, оно же Hitlist size - максимальное количество последовательностей в выдаче.
Short queries - оптимизировать параметры для коротких последовательностей поиска (например, wordsize уменьшается, чтобы увеличить возможное количество слов для инициации выравнивания).
Expect threshold - граничное значение Expect value белка, который мы считаем за попадание. Expect value, оно же E-value - ожидаемое число случайных находок с таким же или выше score базе данных такого же размера для того же белка. К примеру, E-value=1 значит, что в выдачу BLAST данного белка по данной базе данных в среднем может попасть 1 случайный белок, никак не связанный с исходным - просто из-за случайного совпадения последовательностей. Стоит заметить, что E-value само по себе не несёт биологического смысла - это математический параметр, который показывает статистическую значимость данного BLAST-попадания. Чем меньше E-value, тем эта значимость выше. Чем больше база и меньше исходная последовательность, тем (в среднем) выше E-value. В BLAST при расчёте также учитывается неравномерность аминокислотного состава белков (регионы низкой сложности и статистическое распределение по частотам аминокислот).
Word size - минимальная совпадающая последовательность, которая может (во второй фазе поиска) начать выравнивание. Чем меньше значение, тем медленнее, но в некоторых случаях точнее, работает BLAST.
Max matches in a query range - ограничение максимального количества попаданий для участка последовательности (чтобы можно было также увидеть возможное сходство менее консервативных регионов.
Database - UniprotKB/Swiss-Prot(swissprot) (Posted date: Apr 7, 2019 2:39 AM. Number of letters: 177,577,318. Number of sequences: 470,554) - база данных, в которой производится поиск. Причины использования Swiss-Prot - см. далее. Основные базы данных - неизбыточные белковые последовательности (Non-redundant protein sequences (nr)); референсные б.п. (Reference proteins (refseq_protein)); белки модельных организмов (Model Organisms (landmark)); UniProtKB/Swiss-Prot(swissprot); белки с трёхмерной структурой в PDB (Protein Data Bank proteins(pdb)).
Matrix - матрица весов для замен аминокислот. Есть разные матрицы - главное отличие между BLOSUM (blocks substitution) и PAM (point accepted mutation) в способе получения матрицы. BLOSUM получают на основе выравниваний всех последовательностей в базе данных BLOCKS, цифра означает максимальный процент идентичности последовательностей, выравнивания которых учитывались при получении матрицы. Score здесь - вероятность встретить такие аминокислоты сопоставленными друг другу при выравнивании. 0 означает, что эти аминокислоты заменяют друг друга с частотой, приблизительно соответствующей случайной, положительный - чаще, чем ожидалось случайно, отрицательный - наоборот, реже. Модули значений - логарифм отношения встречаемой частоты замены и теоретической (перемноженные частоты встречаемости аминокислот).
Gap opening (existence) cost и Gap exstension cost - уже знакомые нам значения (штраф за открытие гэпа и его продолжение соотвественно).
Compositional adjustments - комплексный набор поправок, который по умолчанию включает в себя и фильтр участков низкой сложности (именно поэтому фильтр * "отключён" - no (default)), и статистическое рапределение частот аминокислот для более точного вычисления E-value и score.
Mask for lookup table only - проводить только первую часть алгоритма BLAST (поиск по словам для построения таблицы) с фильтром на участки низкой сложности и повторы, а вторую (выравнивания) без них.
Mask lower case letters - не учитывать в поиске части последовательности, набранные буквами в нижнем регистре.
Результаты работы BLAST для моего белка:
Оказалось, что гомологов у бета-цепи фенилаланил-тРНК трансферазы Bacteroides fragilis NCTC 9343 более чем достаточно, чтобы заполнить 20000-ную выдачу BLAST при поиске по неизбыточной (nr) (default) и референсной (refseq_protein) базам данных. У всех этих белков одинаковое имя и мнемоника Uniprot (где она есть), а также E-value ниже 6e-130 и 2e-125 соответственно.
В базе SwissProt были найдены (точнее, получены в выдаче за счёт её меньшего размера) более разнообразные белки, например, не только фенилаланил-тРНК синтетаза, но и тирозил-тРНК синтетаза. У этих аминокислот очень похожая структура, поэтому сходство вполне возможно.
Ссылка на скачивание результирующей таблицы BLAST в формате .xlsx
Я выбрала белки наиболее интересных мне организмов (например, Mus musculus, Homo sapiens, Clostridium tetani, Yersinia pestis) из разных таксонов и выровняла их с помощью алгоритма множественного выравнивания muscle. Ссылка на файл с выравниванием - с тирозил-тРНК синтетазой, без неё. Более того, я ещё и использовала BLAST в Uniprot - тоже таблица, получила приблизительно те же результаты (но с 384 hits вместо 325).
У архей и эукариот этот белок похож гораздо больше, чем на бактерий. При этом у бактерий, даже близких видов, между собой последовательности похожи меньше, чем у эукариот. Было найдено 2 достаточно высококонсервативных участка - позиции 187-195, позиции 510-531 (рис. 1), что, как и общая функциональная схожесть, подтверждает гомологию всех рассматриваемых белков. Кроме того, часты единичные (у одного-двух организмов) замены аминокислот (например, позиции 186, 530, 532, 533, 534 на рис. 1), при этом у остальных белков на этой позиции аминокислота одна и та же.
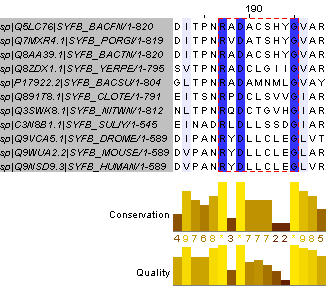
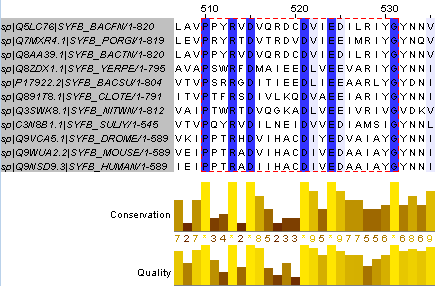
Ссылка на скачивание выравнивания
Задание 2. DotPlot
Программой blastp (единственная изменённая настройка - wordsize=2) было проведено сравнение двух белков: M7XPQ0_RHOT1 Folic acid synthesis protein RHTO_00942 Rhodosporidium toruloides (strain NP11) (Yeast) (Rhodotorula gracilis) (длина 808 а.о.); S8F0X9_FOMPI Uncharacterized protein FOMPIDRAFT_1132301 Fomitopsis pinicola (strain FP-58527) (Brown rot fungus) (длина 258 а.о.). На графике dot-plot (рис. 3) видно практически точное совпадение S8F0X9_FOMPI (10-255) с начальным участком M7XPQ0_RHOT1 (5-303), есть делеции (например, 73-87, 220-241, в основном у более короткого белка, но есть и в большом), а также дальнейшее повторение некоторых участков в первом белке (также совпадающих с соответствующими регионами второго), например, 155-192 M7XPQ0_RHOT1; 729-766 M7XPQ0_RHOT1; 136-173 S8F0X9_FOMPI.
Из графика можно сделать вывод, что скорее всего, второй белок гомологичен начальному участку первого или является фрагментом белковой последовательности. Есть также графическое представление выравниваний (рис. 4) и файл со сравнениями всех участков.
Ссылка на скачивание файла сравнения.
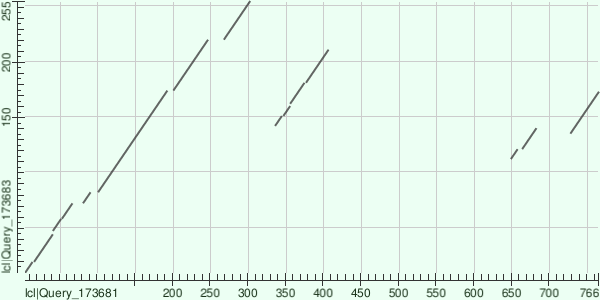

Задание 3.
Для поиска по точно небелковой последовательности я воспользовалась генератором случайной последовательности (написанным мной в 1 семестре). Можно также попытаться с человеческими текстами - если в поисковой последовательности присутствуют буквы, которых в аминокислотном коде нет, то они заменяются, а пробелы и цифры не учитываются (так как они встречаются в формате GenBank). Тем не менее, первые 10 попыток (в основном тексты статей) не принесли результата. Как, впрочем, и случайно сгенерированные варианты длиной 100, 500, 1000, 1500 аминокислот (по 2 попытки). Зато получилось много hits при введении Uniprot AC SYFB_BACFN (преобразованного в SYFBBACFN). В лучшем совпадении E-value была 0.66 с 71.43% идентичности, правда, score 26.9 (что доказывает, что только на E-value и процент идентичности смотреть не стоит). Максимальная E-value (у худшего из совпадений) - 62447. Установление wordsize на 2 и 3 чуть замедлили поиск, но результат не изменился.
Алгоритмы: PHI-BLAST - дополнительно вводится мотив, который обязательно должен присутствовать в результатах; PSI-BLAST - в несколько этапов строится PSSM (position-specific scoring matrix) на основе первой итерации blastp, её также можно редактировать (или вовсе задать свою изначально), и дальше алгоритм работает с ней, а не с, например, BLOSUM. PSSM - это матрица, в которой на score в первую очередь влияют конкретные позиции аминокислот (и, соответственно, их консервативность и функциональная значимость), а не общая похожесть/непохожесть аминокислот в белке и их свойств. Пример участка такой матрицы приведён на рис. 5. DELTA-BLAST - принцип похож на PSI, но PSSM уже известны и построены по базе консервативных доменов (CDD), причём для каждого домена отдельно, что позволяет точнее их распознавать. Для PSI и DELTA также можно выставить порог статистической значимости (по умолчанию 0.005)
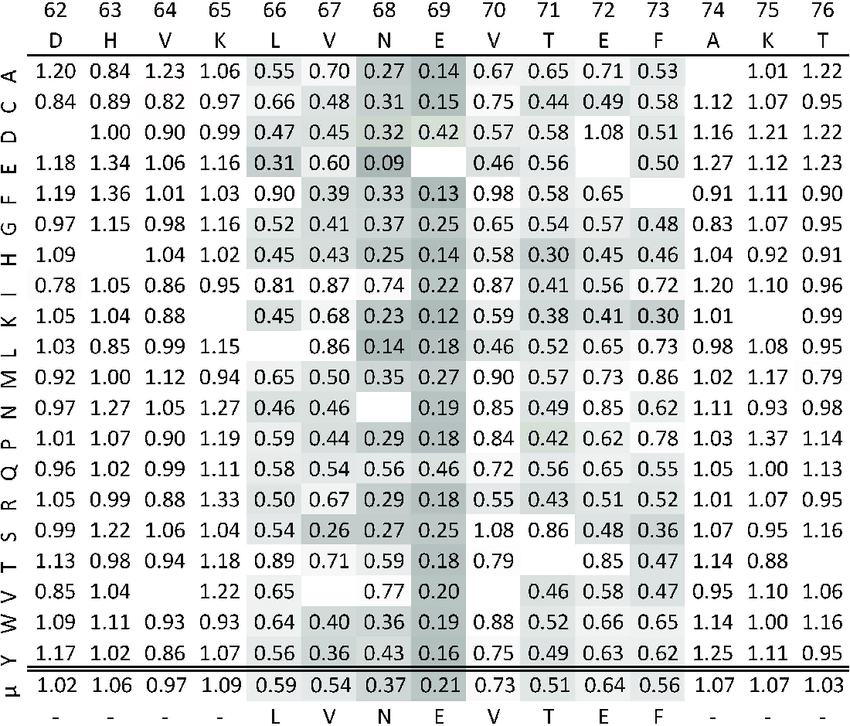
Для PHI-BLAST нужен точно известный паттерн. Я взяла первые 60 букв своего белка, в результате были найдены только 2 белка того же вида бактерий, но разных штаммов. Для моего белка на первой итерации DELTA-BLAST в Swiss-Prot было найдено 724 варианта с максимальной E-value 0.004. Причём на этот раз в список попали также метионил-, тирозил- и пирролизил-тРНК синтетазы, а у многих фенилаланил-тРНК синтетаз, наоборот, статистическая значимость оказалась больше порога. На второй итерации в 1250 найденных последовательностей попали почти все остававшиеся фенилаланил-тРНК синтетазы и несколько лейцин-богатых белков млекопитающих. На третьей в 1232 последовательностей также попали гистидил- и O-фосфосерил-тРНК синтетазы. Для правильной работы PSI-BLAST стоит корректировать белки, на которых рассчитывается матрица, и саму матрицу на каждом шаге (оставляя только точно известные совпадения).
Опция Exclude: Exclude models (XM/XP accessions) - исключить белки, полученные трансляцией предполагаемых (не подтверждённых) кодирующих последовательностей из RefSeq; Non-redundant RefSeq proteins (WP) - исключить вообще все белки RefSeq (не уверена); Uncultured/environmental sample sequences - исключить загрязняющие последовательности (с неподтверждённым источником-организмом).
Для SYFB и ATPA никакие опции Exclude не изменили результата (в поиске по nr, в Swiss-prot они бесполезны из-за состава самой базы). Возможно, стоило попробовать менее распространённые белки.
Ссылки на источники
- BlastHelp – документация по использованию BLAST.
- researchgate.net/publication/312515756_ArrayPitope_Automated_Analysis... – статья, из которой взят пример PSSM.