Практикум 10. Геномные браузеры
Для анализа мной был выбран ген TERT (для человека также встречается обозначение hTERT), кодирующий белок telomerase reverse transcriptase. Он удлиняет теломеры в стволовых, эмбриональных и раковых клетках человека, позволяя им делиться без сокращения длины теломер, в нормальных дифференцированных клетках неактивен. Является онкогеном.
Задание 1. UCSC
- Имя гена (короткое): TERT
- Идентификатор гена в Gencode: ENSG00000164362.20
- Цепь: -
- Хромосома: 5
- Регион: chr5:p15.33
- Число альтернативных транскриптов: 2 (3)
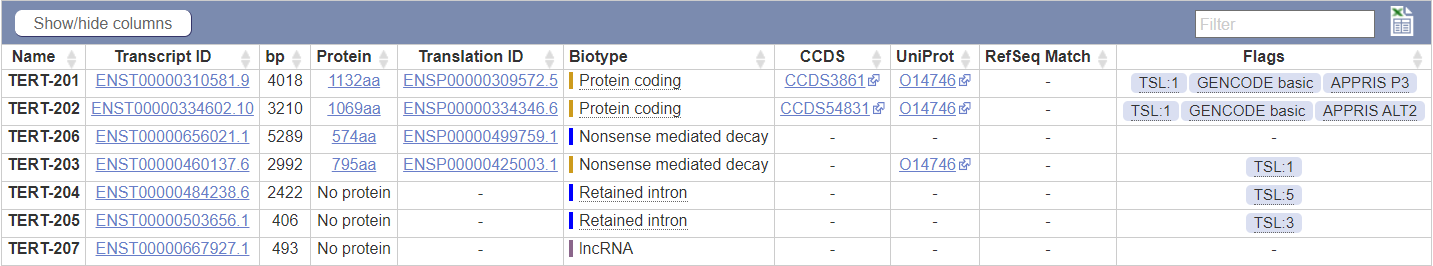
В RefSeq указано 4 транскрипта и 4 изоформы белка, в Ensembl 7 транскриптов, в то время как в GenCode всего 2, каждый кодирует отдельную изоформу белка. Есть также третий отображаемый транскрипт, но он не относится к TERT (ген AC114291.2). Также, он не кодирует белок и находится на обратной цепи. Не описан.
| Характеристика | Транскрипт 1 | Транскрипт 2 |
|---|---|---|
| Идентификатор Gencode | ENST00000310581.9 | ENST00000334602.10 |
| Координаты в хромосоме (включая UTR) | chr5:1,253,167-1,295,047 | chr5:1,253,728-1,294,989 |
| Число экзонов (общее) | 16 | 15 |
| Длина последовательности белка | 1132 aa | 1069 aa |

Лучше открывать картинку в полном размере.
Задание 2. Ensembl
В Ensembl, как упоминалось, нашлось 7 транскриптов, два из них кодируют изоформы работающего белка, ещё две - неработающий (nonsense) белковый продукт, оставшиеся три вообще не транслируются.
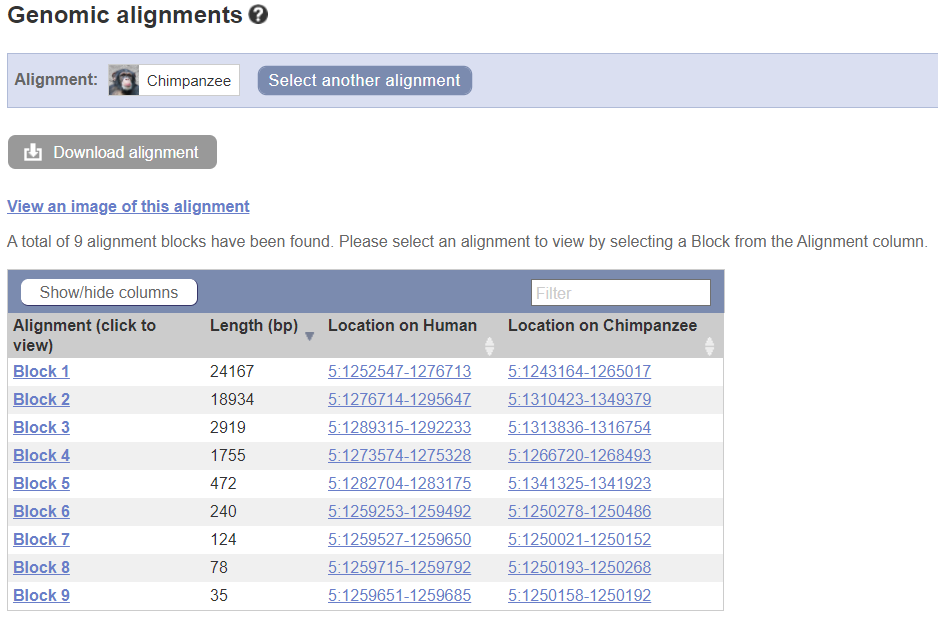
При выравнивании генов человека и шимпанзе обыкновенного (Pan troglodytes) выяснилось, что одного полного выравнивания нет - оно представлено блоками. Судя по координатам в геноме, у шимпанзе произошла инсерция участка 5:1265017-1310423 (либо у человека делеция соответствующего) - рис. 3. Для уточнения я обратилась к выравниваниям с шимпанзе бонобо (Pan paniscus) и западной гориллой (Gorilla gorilla). Для других гоминид выравнивание полное, так что скорее всего, произошла инсерция у шимпанзе.
Так что для анализа я решила использовать выравнивание TERT человека с TERT бонобо. Как видно на рис. 4, предлагаемые блоки - составные части основного выравнивания (верхнего).
Скачать выравнивание TERT человека и бонобо в формате fasta.
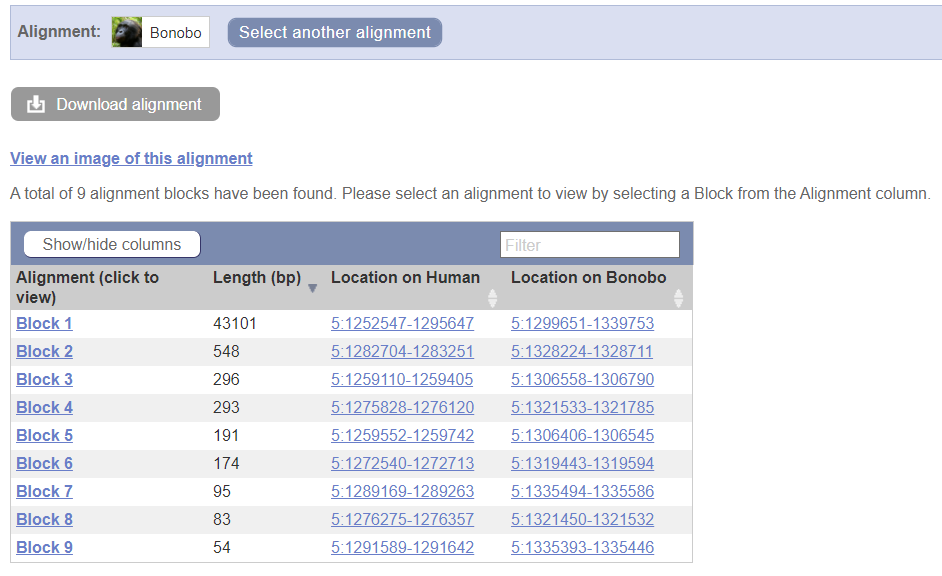
Общее различие в геномах человека и бонобо - 1.3% (Ссылка на статью)
В этом выравнивании я посчитала характеристики командой infoalign, синтаксис: infoalign -only -name -seqlength -gaps -idcount -diffcount -heading -html XXX.fasta (результат представлен в таблице ниже).
Результат: процент различия = (1 - (36148/37490)) * 100% = 3.58%, что в 2.75 раза больше, чем в среднем по геному. В этой формуле не учитываются гэпы, она позволяет установить только различия в последовательности.
| Sequence Length | Gaps | Identity | Difference |
|---|---|---|---|
| 43101 | 50 | 43101 | 0 |
| 37490 | 74 | 36148 | 1342 |
Задание 3. Genome Data Viewer
В поле External references Ensembl есть ссылка на NCBI.
Ссылка на External references Ensembl для TERT
Ссылка на Genome Data Viewer для TERT
В целом,сделать выводы по количеству предоставляемой информации сложно. И в браузере UCSC, и в GDV есть большое количество дополнительных треков, которые выдают практически всю известную научному сообществу информацию об анализируемом участке генома.
Из предоставляемой по умолчанию информации я нашла в Genome Data Viewer интересные пункты, представленные ниже. Но в нём есть различная дополнительная информация и множество треков, которые доступны в меню. Я бы сказала, что графическая репрезентация в GDV удобнее и интерактивнее, чем в UCSC, но это скорее связано с тем, что с ним я уже работала.
Стоит также заметить, что Genome Data Viewer позволяет сразу пользоваться инструментами баз данных NCBI - переходить в нужную базу данных или статью Pubmed при наведении мышкой, перейти к FASTA-последовательности из RefSeq/GenBank и проводить BLAST для отмеченных (а маркеры можно ставить вручную) участков, что во многих случаях очень удобно.
Name: RNA-seq exon coverage, aggregate (filtered), NCBI Homo sapiens Annotation Release 109 Comment: Exon coverage of RNA-seq alignments, filtered to remove low abundance alignments and some apparent retained-intron alignments near splice junctions and scaled with a linear scaled transform.
Name: RNA-seq intron-spanning reads, aggregate (filtered), NCBI Homo sapiens Annotation Release 109 Comment: Coverage of introns derived from spliced RNA-seq alignments, filtered to remove low abundance alignments and some apparent retained-intron alignments near splice junctions and scaled with a linear scaled transform.
