Практикум 11
Команды для работы с чтениями
Анализируемая хромосома: 20
| Команда | Функция |
|---|---|
| hisat2-build ${chr_file} ${chr_name}_index 1>${chr_name}_hisat2_index.log 2> ${chr_name}_hisat2_index.txt | Проиндексировать референсную последовательность хромосомы |
| fastqc ${chr_reads} 2> ${chr_name}_reads_fastqc.log | Анализ качества чтений до очистки |
| ${trimmomatic} SE -phred33 ${chr_reads} ${chr_name}_trimmed.fastq TRAILING:${trim_trailing} MINLEN:${trim_minlen} 2> ${chr_name}_trimmomatic.log | Очистка чтений |
| fastqc ${chr_reads_trim} 2> ${chr_name}_reads_trim_fastqc.log | Анализ качества чтений после очистки |
| hisat2 -x ${chr_index} -U ${chr_reads_trim} -S ${chr_sam} --no-spliced-alignment --no-softclip 2> ${chr_name}_hisat2_align.log | Картирование чтений на проиндексированную референсную последовательность, выравнивание в формате sam |
| samtools view -b -o ${chr_bam} ${chr_sam} 2> ${chr_name}_samtools_align_view.log | Перевод выравнивания чтений с референсом из формата sam в формат bam |
| samtools sort ${chr_bam} ${chr_bam_sort} 2> ${chr_name}_samtools_align_sort.log | Сортировка выровненных чтений в файле bam по координате |
| samtools index ${chr_bam_sort}.bam 2> ${chr_name}_samtools_align_index.log | Индексирование отсортированных выровненных чтений |
| samtools flagstat ${chr_bam_sort}.bam > ${chr_name}_flagstat.txt | Выдача информации о картировании в файл, в т.ч. числа откартированных чтений |
| samtools mpileup -u -f ${chr_file} ${chr_bam_sort}.bam -o ${chr_name}_SNPaI.bcf 2> ${chr_name}_samtools_mpileup.log | Создание файла bcf со списком полиморфизмов, включая индели |
| bcftools call -cv ${chr_name}_SNPaI.bcf -o ${chr_name}_SNPaI.vcf 2> ${chr_name}_bcftools_call.log | Перевод файла из формата bcf в формат vcf |
| vcftools --vcf ${chr_name}_SNPaI.vcf --remove-indels --recode --out ${chr_name}_SNP 2> ${chr_name}_vcftools_SNP.log | Удаление инделей из файла, оставляя только SNP |
| convert2annovar.pl -format vcf4 ${chr_name}_SNP.recode.vcf > ${chr_name}_SNP.avinput 2> ${chr_name}_convert2annovar.log | Получение входного файла для annovar |
| ${annovar} -out ${chr_name}_refgene -build hg19 ${chr_name}_SNP.avinput ${humandb_dir} | Аннотация полиморфизмов с использованием базы данных refgene |
| ${annovar} -filter -out ${chr_name}_dbsnp -build hg19 -dbtype snp138 ${chr_name}_SNP.avinput ${humandb_dir} | Аннотация полиморфизмов с использованием базы данных dbsnp |
| ${annovar} -filter -out ${chr_name}_1000g -build hg19 -dbtype 1000g2014oct_all ${chr_name}_SNP.avinput ${humandb_dir} | Аннотация полиморфизмов с использованием базы данных 1000 genomes |
| ${annovar} -regionanno -out ${chr_name}_gwas -build hg19 -dbtype gwasCatalog ${chr_name}_SNP.avinput ${humandb_dir} | Аннотация полиморфизмов с использованием базы данных Gwas |
| ${annovar} -filter -out ${chr_name}_clinvar -build hg19 -dbtype clinvar_20150629 ${chr_name}_SNP.avinput ${humandb_dir} | Аннотация полиморфизмов с использованием базы данных Clinvar |
Анализ качества чтений до и после очистки
Всего было 4661 чтений, их качество (из выдачи fastqc) показано на рисунке 1. После очистки чтений trimmomatic - убраны нуклеотиды с качеством менее 20, минимальная длина для чтений после этой обработки 50 - осталось 4472 (95,95%), были отброшены 189 (4,05%). Их качество (из выдачи fastqc) показано на рисунке 1. Судя по результатам, триммирование оправдано - отброшены немного чтений, но среднее качество по последовательности, особенно в конце, значительно выросло. Одна из любопытных характеристик, приведённых в отчёте fastqc - per sequence GC content. Теоретически, распределение должно быть нормальным, но у меня что до триммирования, что после, нормального распределения не получалось. Почему - хороший вопрос. Пример приведён на рисунке 3.
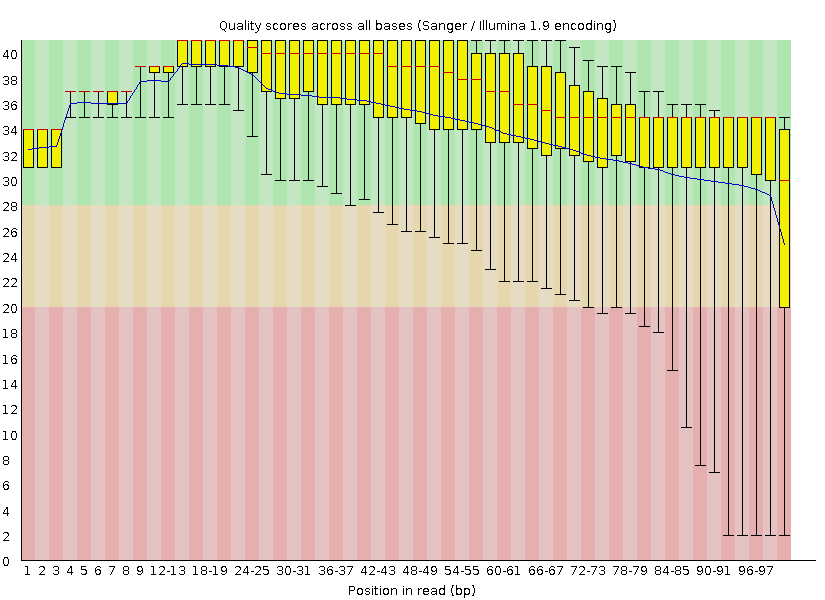
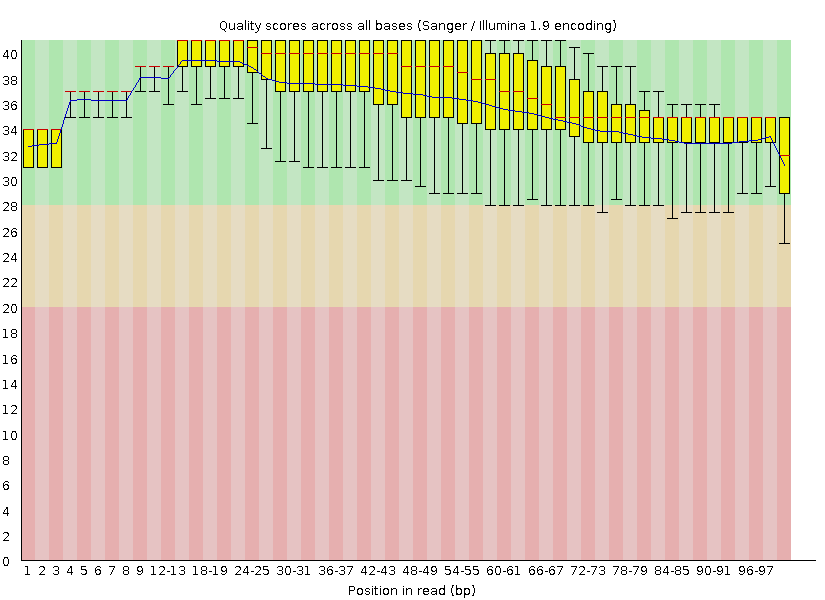
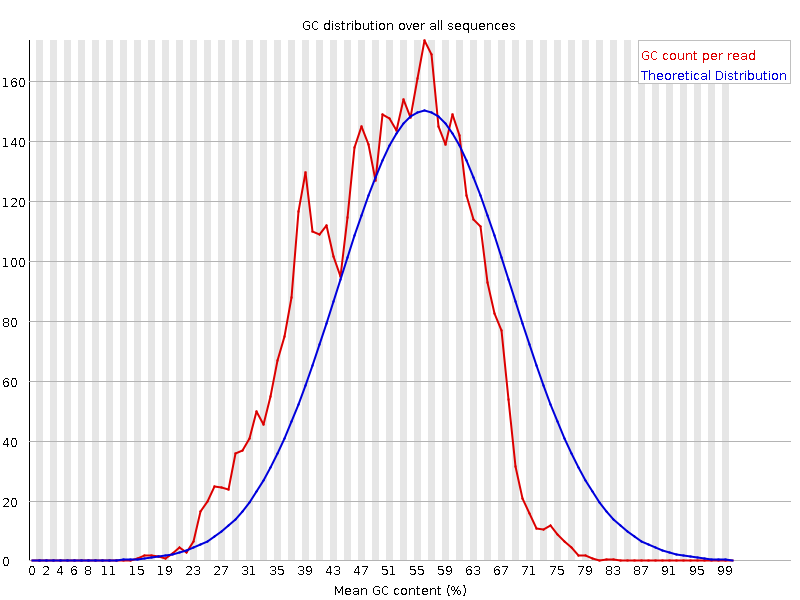
Картирование чтений
Из 4472 чтений 17 не удалось выровнять на референс, 4454 выровнялись ровно один раз, 1 выровнялось 2 раза (общий процент выравнивания 99.62%).
Описание полиморфизмов
Всего полиморфизмов 42, все они одного типа - замена (нет делеций и вставок), причём полиморфизмы сконцентрированы на трёх участках. Большинство, тем не менее, имеют очень низкое покрытие (1-2 чтения) и качество - возможно, ошибки секвенирования. 16 из 42 имеют хорошее покрытие. Описания трёх полиморфизмов с хорошим покрытием приведены в таблице 2.
| Номер | Координата | Тип полиморфизма | Референс | Альтернатива (чтения) | Глубина покрытия | Качество чтений |
|---|---|---|---|---|---|---|
| 1 | 33974207 | Замена | A | G | 39 | 207.01 |
| 2 | 48524827 | Замена | A | G | 40 | 225.01 |
| 3 | 56190634 | Замена | C | T | 37 | 225.01 |
Аннотация полиморфизмов
База данных refseq даёт о полиморфизмах следующую информацию: в координатах какого гена (или координаты области полиморфизма относительно соседних генов) находится полиморфизм, в интронной, экзонной, нетранслируемой или междугенной области. Из 42 полиморфизмов 15 находятся в интронных областях гена UQCC1 (Ubiquinol-Cytochrome C Reductase Complex Assembly Factor 1), 3 между генами UQCC1 и GDF5, 1 в экзоне GDF5 (Growth differentiation factor 5), по одному в UTR5 и upstream областях того же гена, 3 в экзонах гена SPATA2 (Spermatogenesis Associated 2), 1 в upstream и 2 в интронах того же гена, 4 в экзонах ZBP1 (Z-DNA-binding protein) и 11 в его интронах.
Из экзонных замен 4 являются синонимичными - 3 в гене SPATA2 (exon3:c.C1389T:p.C463C; exon3:c.C1134T:p.S378S; exon2:c.T201C:p.Y67Y), 1 в гене ZBP1 (exon7:c.C1086T:p.D362D).
Несинонимичные замены: 1 в GDF5 (exon2:c.T826G:p.S276A), 3 в ZBP1 (exon6:c.A770G:p.Q257R; exon4:c.G460C:p.D154H; exon3:c.G262A:p.E88K)
Из 42 полиморфизмов 32 имеют reference SNP ID number (rs).
12 полиморфизмов встречается довольно часто (диапазон 0.35-0.6), 3 реже (0.13-0.19) 13 очень редко (менее 0.1), есть несколько очень распространённых (0.695, 0.813 и даже 0.996).
Cогласно базе gwas, три из найденных полиморфизмов отвечают за рост, псориаз и атрофию гиппокампа соответственно.
В clinvar есть запись только об одном из найденных полиморфизмов - в гене GDF5, он ведёт к повышенному риску возникновения остеоартроза бедренного сустава (CLNACC=RCV000008898.2).
Визуализация полиморфизмов программой IGV
У меня, к сожалению, не получилась - IGV просто не показывал отсортированные чтения, хотя все остальные инструменты с ними работали нормально. FAQ утверждает, что это может быть связано с неправильным именованием хромосом в нашем файле.