Практикум 12. RNA-seq
Команды для работы с чтениями
Анализируемая хромосома: 20
| Команда | Функция |
|---|---|
| hisat2-build ${chr_file} ${chr_name}_index 1>${chr_name}_hisat2_index.log 2> ${chr_name}_hisat2_index.txt | Проиндексировать референсную последовательность хромосомы |
| fastqc ${chr_rna_reads} 2> ${chr_name}_rna_reads_fastqc.log | Анализ качества чтений до очистки |
| ${trimmomatic} SE -phred33 ${chr_rna_reads} ${chr_name}_rna_trimmed.fastq TRAILING:${trim_trailing} MINLEN:${trim_minlen} 2> ${chr_name}_rna_trimmomatic.log | Очистка чтений |
| fastqc ${chr_name}_rna_trimmed.fastq 2> ${chr_name}_rna_reads_trim_fastqc.log | Анализ качества чтений после очистки |
| hisat2 -x ${chr_index} -U ${chr_name}_rna_trimmed.fastq -S ${chr_sam} --no-softclip 2> ${chr_name}_rna_hisat2_align.log | Картирование чтений на проиндексированную референсную последовательность, выравнивание в формате sam. По сравнению с практикумом 11 убрала опцию --no-spliced-alignment - в мРНК вполне может быть сплайсинг |
| samtools view -b -o ${chr_bam} ${chr_sam} 2> ${chr_name}_rna_samtools_align_view.log | Перевод выравнивания чтений с референсом из формата sam в формат bam |
| samtools sort ${chr_bam} ${chr_bam_sort} 2> ${chr_name}_rna_samtools_align_sort.log | Сортировка выровненных чтений в файле bam по координате |
| samtools index ${chr_bam_sort}_rna.bam 2> ${chr_name}_rna_samtools_align_index.log | Индексирование отсортированных выровненных чтений |
| htseq-count -f bam ${chr_bam_sort}_rna.bam -i gene_id -s no gencode.v19.chr_patch_hapl_scaff.annotation.gtf > counts_all.txt | Подсчёт всех чтений, включая результаты с нулевым числом чтений |
| grep -wv "0" counts_all.txt > counts_nozero.txt | Выдача из предыдущего файла только строк, в которых упоминается ненулевое количество чтений |
Первая реплика
Анализ качества чтений до и после очистки
Всего было 3565 чтений, их качество (из выдачи fastqc) показано на рисунке 1. После очистки чтений trimmomatic - убраны нуклеотиды с качеством менее 20, минимальная длина для чтений после этой обработки 50 - осталось 3530 (99,02%) 35 (0,98%), были отброшены 35 (0,98%).
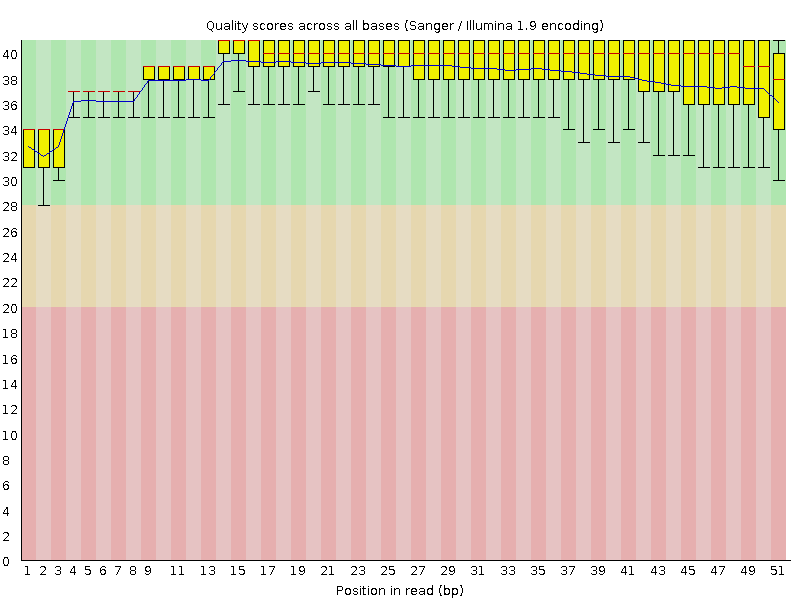
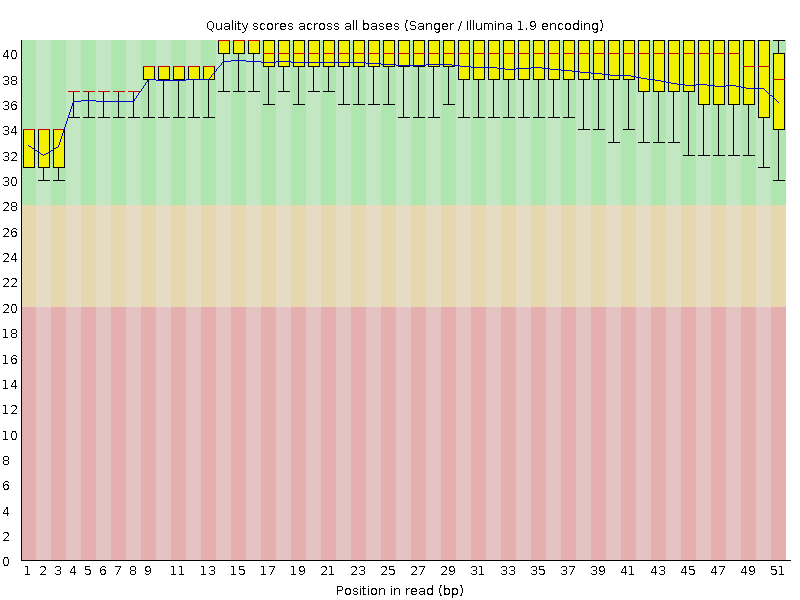
Подсчёт чтений
Из 3565 чтений легло на ген ENSG00000125835.13 - 1702, на ген ENSG00000251806.1 - 4, вне обозначенных разметкой участков (no_feature) - 274, неоднозначно (ambiguous) - 633, не выровнялось вовсе (not_aligned) - 917. Я бы сказала, что выровнялось довольно плохо - почти половина чтений либо не выровнены, либо выровнены неоднозначно. Низкокачественно (too_low_aQual) и неуникально (alignment_not_unique) выровненных чтений не было - 0.
Гены
ENSG00000125835.13 - ген, кодирующий полипептиды B и B1 малых ядерных рибонуклеопротеинов (small nuclear ribonucleoprotein polypeptides B and B1). Малые ядерные рибонуклеопротеины (snRNP) участвуют в сплайсинге мРНК. ENSG00000251806.1 - ген малой ядрышковой РНК (small nucleolar RNA, C/D box 119). Функцию конкретно её никто не знает, но в целом такие snoRNA направляют другие РНК.