
Упражнение 4.

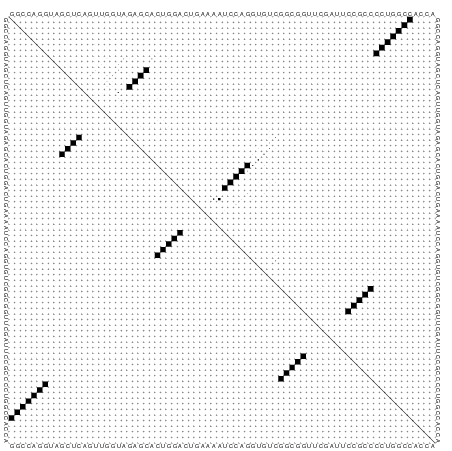
einverted trna.fastaПри запуске программы с параметрами по умолчанию в выходные файлы s.inv и s.fasta ничего не было записано. После понижения значения параметра minimum score threshold до 40 в файлы ничего не было записано. После понижения значения параметра minimum score threshold до 40 в файлы ничего не было записано. После понижения значения параметра minimum score threshold до 20 в файлы f.inv и f.fasta была записана информация, но ее оказалось очень мало. Для того, чтобы увеличить количество данных, записываемых в файлы, были смягчены критерии, по которым программа отбрасывает информацию. Чтобы score threshold был больше был увеличен Match score(с 3 до 5), снижен по модулю Mismatch score(с -4 до -1) и снижен Gap penalty(с 12 до 5). Команда einverted trna.fasta была запущена со следующими параметрами:
Find inverted repeats in nucleotide sequences Gap penalty [12]: 5 Minimum score threshold [50]: 20 Match score [3]: 5 Mismatch score [-4]: -1 Sanger Centre program inverted output file [sequence.inv]: t.inv File for sequence of regions of inverted repeats. [sequence.fasta]: t.fastaВ результате были получены файлы:t.inv и t.fasta. В начале были введены следующие команды, что бы указать путь к RnaFold:
tsvetcovroman@kodomo:~/public_html/term3/block1/pr3$ export PATH=${PATH}:/home/pre
Затем была запущена программа RnaFold:
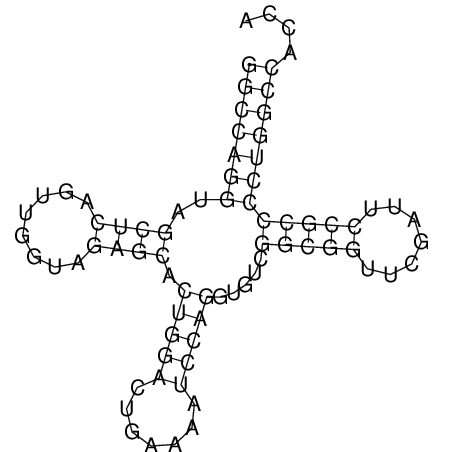
cat trna.fasta | RNAfold --MEA > rna_fold.fastaВыходной файл:rna_fold.fasta, в котором первую строчку представляет собой последовательность, а следующие строчки — предсказанная структура MFE. Квадратными совпадающими скобками обозначена связь пары оснований, и далее указана свободная энергия в ккал/моль. Точка в обозначает нуклеотид, не образующий пару. Кроме того с помощью той же программы были получены файлы 5HC9:C|PDBID|CHAIN|SEQUENCE_dp.ps, 5HC9:C|PDBID|CHAIN|SEQUENCE_ss.ps, 5HC9:D|PDBID|CHAIN|SEQUENCE_dp.ps и 5HC9:D|PDBID|CHAIN|SEQUENCE_ss.ps, которые затем были конвертированы в формат jpeg и представлены на рис. 1,2, 3 и 4 соответственно. Сравнение результатов с выдачей программы find_pair представлены в табл. 1.





| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель |
5' ----------------> 3' 1 (0.001) ....>C:...1_:[..G]G-----C[..C]:..72_:C<.... (0.003) | 2 (0.002) ....>C:...2_:[..G]G-----C[..C]:..71_:C<.... (0.001) | 3 (0.001) ....>C:...3_:[..C]C-----G[..G]:..70_:C<.... (0.002) | 5 (0.002) ....>C:...5_:[..A]A-----U[..U]:..68_:C<.... (0.001) | 6 (0.003) ....>C:...6_:[..G]G-----C[..C]:..67_:C<.... (0.002) | 7 (0.001) ....>C:...7_:[..G]G-----C[..C]:..66_:C<.... (0.002) | Всего 7 пар | 0 верных пар | 7 |
| D-стебель |
8 (0.003) ....>C:..49_:[..G]G-----C[..C]:..65_:C<.... (0.003) 9 (0.001) ....>C:..50_:[..G]G-----C[..C]:..64_:C<.... (0.001) 10 (0.002) ....>C:..51_:[..C]C-*---G[..G]:..63_:C<.... (0.001) 11 (0.005) ....>C:..52_:[..G]G-----C[..C]:..62_:C<.... (0.002) 12 (0.002) ....>C:..53_:[..G]G-----C[..C]:..61_:C<.... (0.002) Всего 5 пар | 0 верных пар | 4 |
| T-стебель |
15 (0.002) ....>C:..38_:[..A]A-**--C[..C]:..32_:C<.... (0.002) | 16 (0.003) ....>C:..39_:[..U]U-*---A[..A]:..31_:C<.... (0.001) | 17 (0.002) ....>C:..40_:[..C]C-----G[..G]:..30_:C<.... (0.001) | 18 (0.002) ....>C:..41_:[..C]C-----G[..G]:..29_:C<.... (0.001) | 19 (0.004) ....>C:..42_:[..A]A-----U[..U]:..28_:C<.... (0.002) | 20 (0.002) ....>C:..43_:[..G]G-**--C[..C]:..27_:C<.... (0.002) | 21 (0.002) ....>C:..44_:[..G]G-**--A[..A]:..26_:C<.... (0.002) | Всего 7 пар | 0 верных пар | 4 |
| Антикодоновый стебель |
22 (0.004) ....>C:..10_:[..G]G-----C[..C]:..25_:C<.... (0.002) | 23 (0.002) ....>C:..11_:[..C]C-----G[..G]:..24_:C<.... (0.004) | 24 (0.003) ....>C:..12_:[..U]U-----A[..A]:..23_:C<.... (0.005) | 25 (0.003) ....>C:..13_:[..C]C-----G[..G]:..22_:C<.... (0.003) | 26 (0.003) ....>C:..14_:[..A]A-**--U[..U]:...8_:C<.... (0.001) | 27 (0.002) ....>C:..15_:[..G]G-**+-C[..C]:..48_:C<.... (0.001) x 28 (0.002) ....>C:..19_:[..G]G-----C[..C]:..56_:C<.... (0.003) + Всего 7 пар | 0 верных пар | 5 |
| Общее число канонических пар нуклеотидов | 26 пар | 0 верных пар | 20 верных пар |
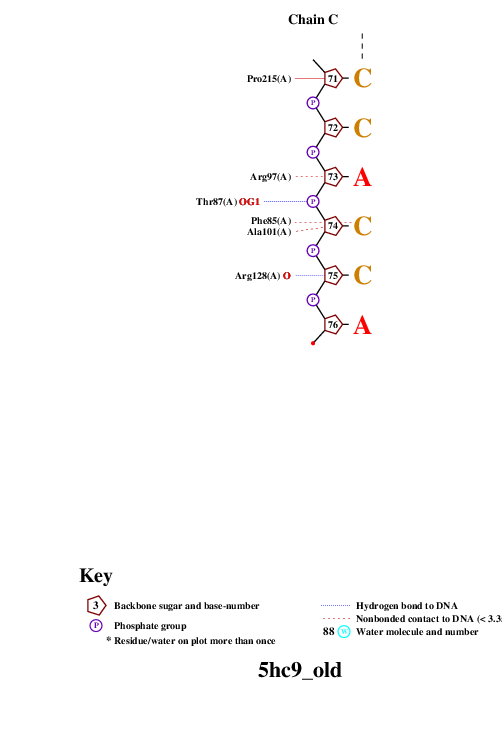
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| Остатками 2'-дезоксирибозы | 11 | 27 | 38 |
| Остатками фосфорной кислоты | 10 | 11 | 21 |
| Остатками азотистых оснований со стороны большой бороздки | 10 | 33 | 43 |
| Остатками азотистых оснований со стороны малой бороздки | 8 | 17 | 25 |
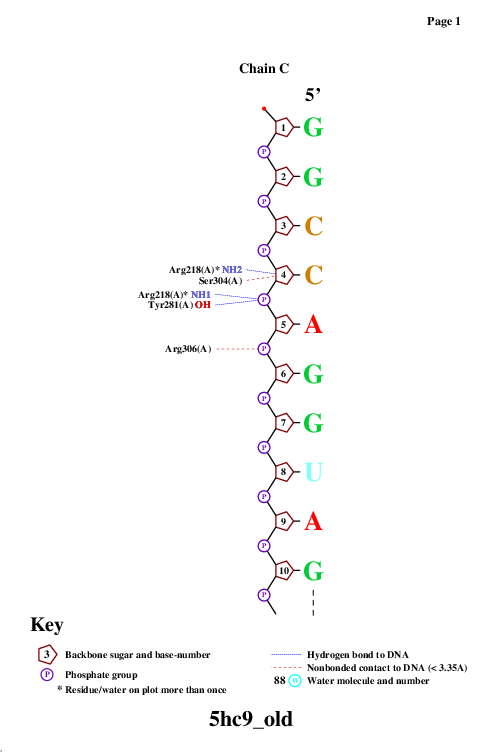
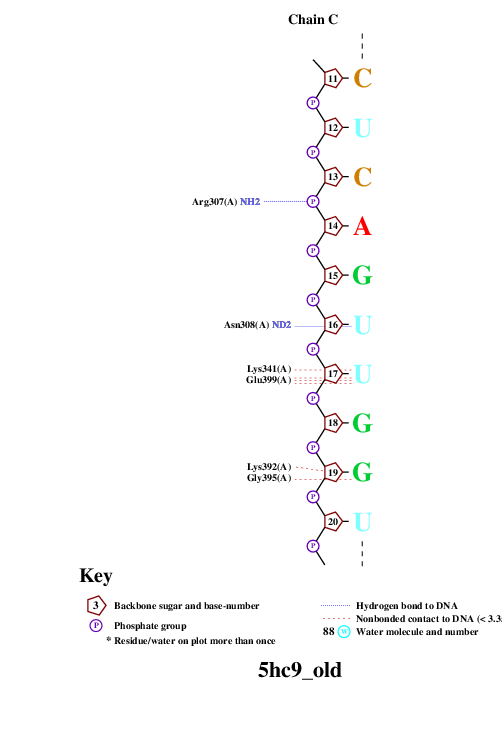
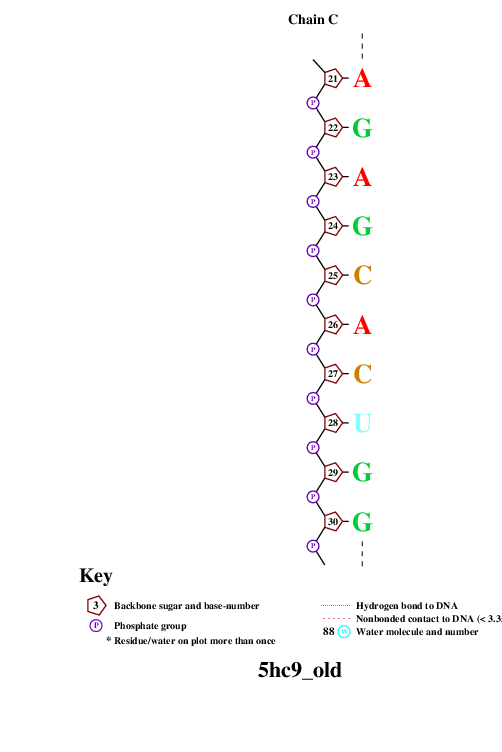
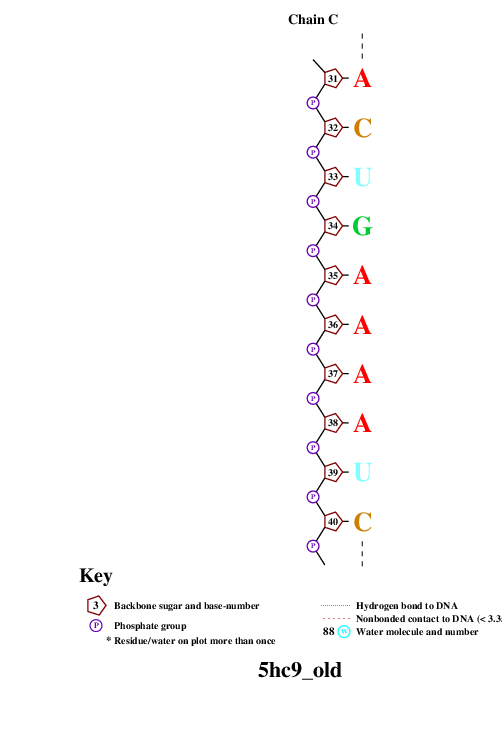
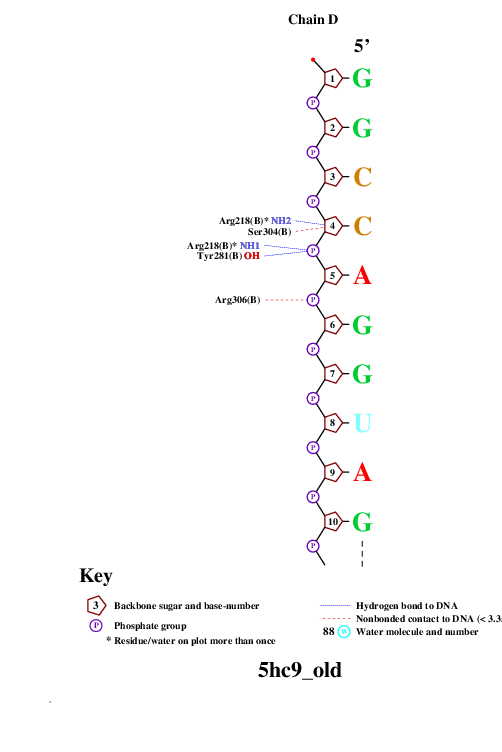
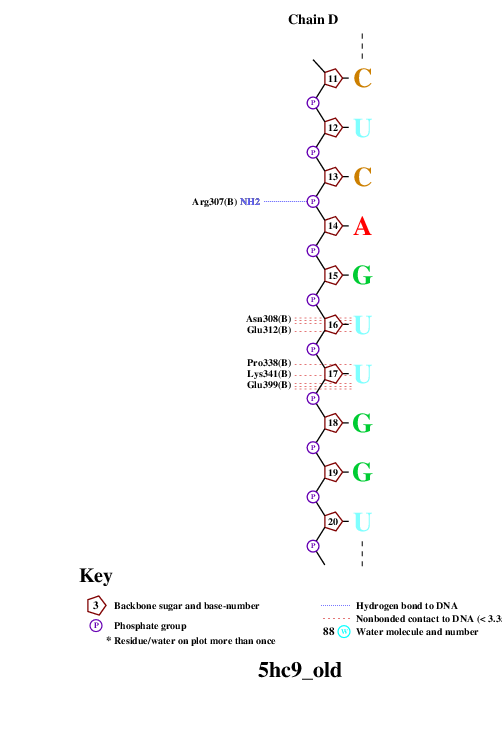
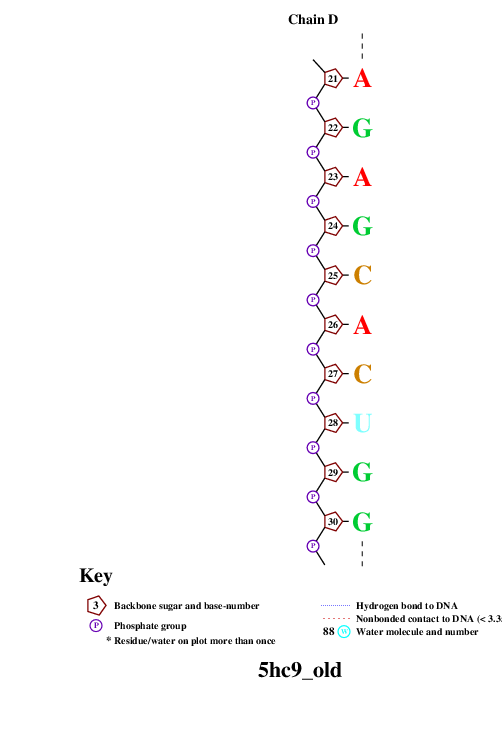
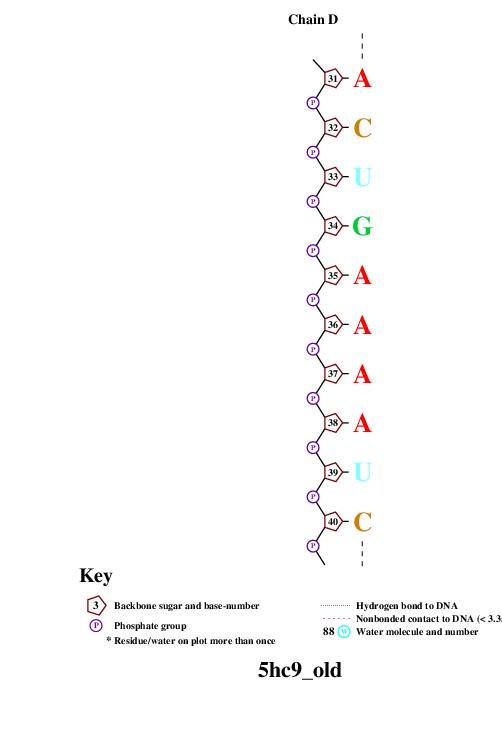
remediator --pdb --old 5hc9.pdb > 5hc9_old.pdbИспользованная для запуска nucplot команда:
nucplot 5hc9_old.pdbНа выходе получали файл-изображение nucplot.ps, который для удобства нужно было конвертировать в [png]-формат:
convert PS:nucplot.ps PNG:nucplot.pngВыдачей программы convert послужили 15 файлов: nucplot-0.png, ..., nucplot-15.png. Они приведены на Рис.2.