Отчет по практикуму 9. EMBOSS. Выравнивание геномов.
На этой странице выложен отчет по практикуму 9. EMBOSS. Выравнивание геномов.
Задание 1.
1. С помощью программы seqret файлы a.fasta , b.fasta , c.fasta , названия которых были записаны в файле
mylist.txt
были объединены в файл d.fasta .
При этом использовалась команда:
seqret @mylist.txt d.fasta
2. С помощью программы seqretsplit файл e.fasta с 4 последовательностями в формате fasta был разделен на 4 отдельных файла, в каждом из которых 1
последовательность в формате fasta:hq608513.1.fasta , kc581915.1.fasta ,
lc036322.1.fasta , nc_020099.1.fasta .
При этом использовалась команда:
seqretsplit e.fasta
3.Из файла с хромосомой s.gb были вырезаны три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранены в одном fasta файле.
Для этого был создан список list.txt, в котором записаны координаты кодирующих последовательностей. С помощью команды:
seqret @list.txt seq.fasta
были вырезаны соответствующие кодирующие последовательности по указанным координатам, каждая записана в файл: seq.fasta.
4. С помощью программы transeq кодирующие последовательности, лежащие в файлеh.fasta , транслированы в аминокислотные, используя указанную таблицу
генетического кода. Для трансляции использовалась таблица генетического кода под номером 11. При этом использовалась команда:
transeq -table 11 h.fasta
Файл с полученными аминокислотными последовательностями:hq608513.pep.
5. С помощью опции frame 6 программы transeq последовательность b.fasta была транслирована в 6 рамках.
При этом использовалась команда:
transeq b.fasta t.fasta -frame 6
В результате
получен файл t.fasta .
6. С помощью программы seqret файл sanger.mfa в формате mfa(fasta) был переведен в формат msf.
Для этого была использована команда seqret sanger.mfa msf::sanger.msf
. В результате получен файл sanger.msf.
7. С помощью программы infoalign и ее опций -refseq 2 и -only -name -idcount из выравнивания
был получен файл a.txt с числом совпадающих букв между второй последовательностью выравнивания и всеми остальными. Для этого использовалась команда:
infoalign alignment.msf a.txt -refseq 2 -only -name -idcount
С помощью команды
cat a.txt
он был выдан в выходной поток.
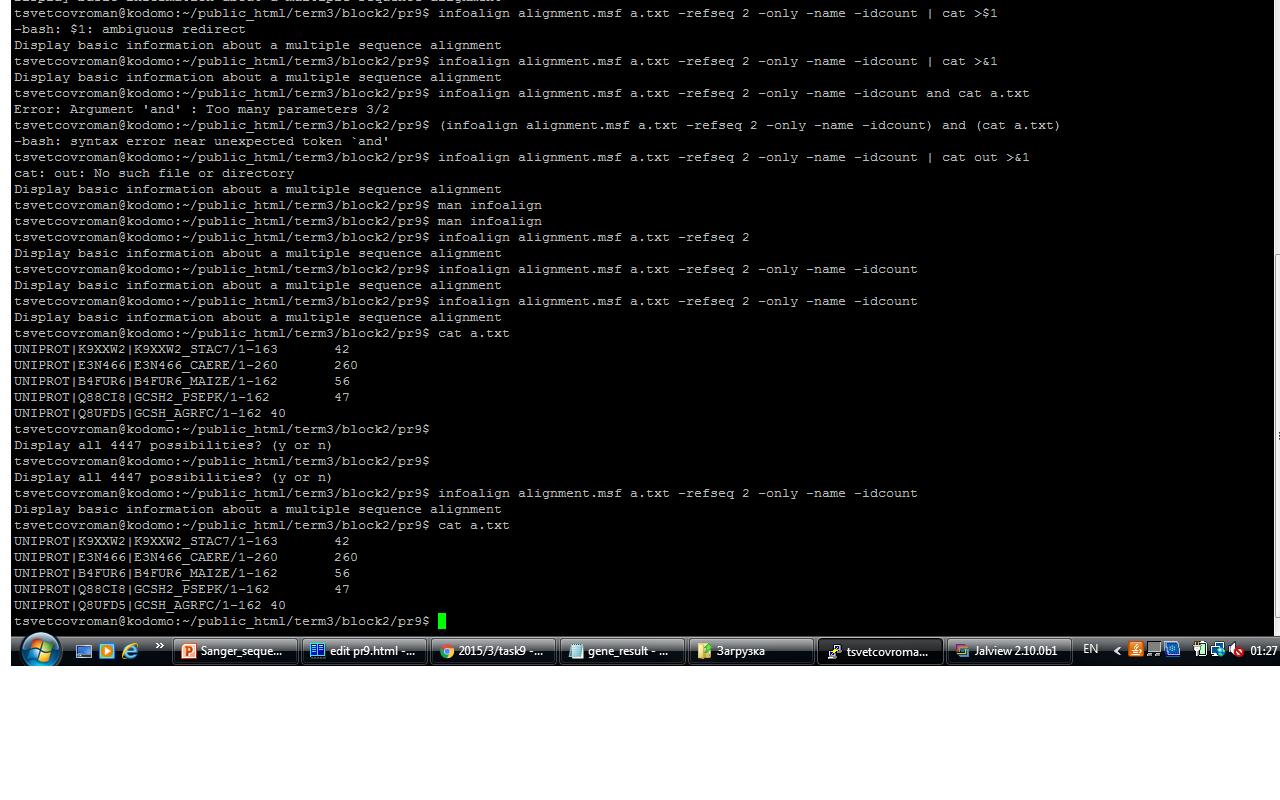
Рисунок 1. Скрин выходного потока после применения команды.
8. С помощью программы featcopy переведены аннотации особенностей в записи формата .gb в табличный формат .gff.
На вход подавалась нуклеотидная последовательность sequence.gb.
featcopy sequence.gb -auto
В результате чего был получен файл
с названием по умолчанию: sequence.gff, содержащий аннотации особенностей в формате таблицы.
9. С помощью extractfeat из одного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями;
добавить в описание каждой последовательности функцию белка (из поля product). На вход опять подавался файл sequence.gb. Использовалась
команда:
extractfeat sequence.gb info.fasta -type CDS -describe product
На выходе был получен файл info.fasta со всеми кодирующими последовательностями из входного файла,
а также с описаниями функции каждого белка, например, (product="hypothetical protein").
10. С помощью программы shuffleseq перемешаны буквы в данной нуклеотидной последовательности s.fasta .
Для этого использовалась команда:
shuffleseq s.fasta r.fasta
В результате получен файл r.fasta .
Далее предлагалось проверить найдет ли blastn достоверные (e-value до 0.1) сходные последовательности в нуклеотидном банке данных.
Для этого был осуществлен поиск blastn с параметрами по умолчанию(порог E-value был взят 10). На рисунке 1 приведены результаты поиска.
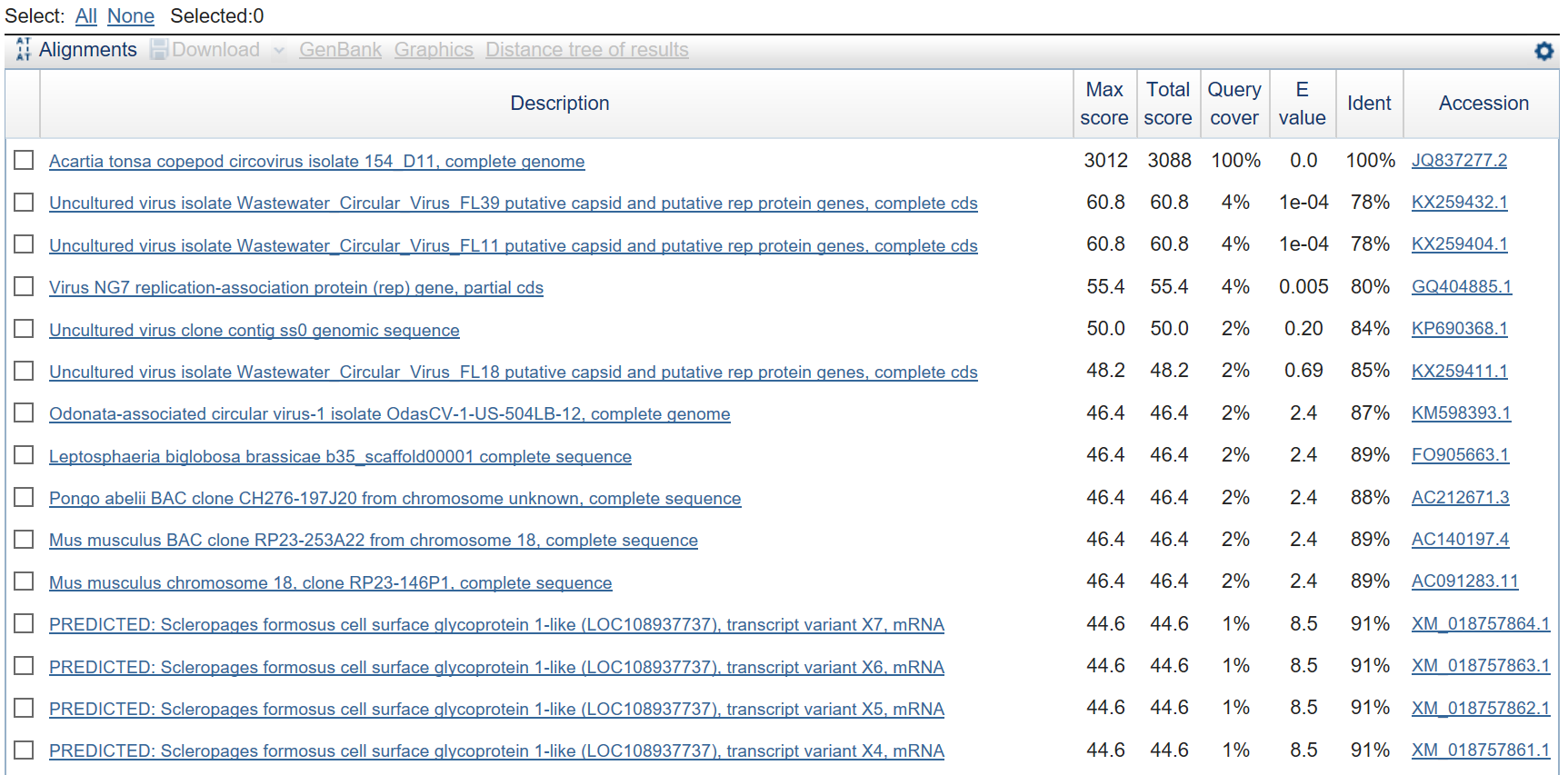 Рис. 1. Выдача Blast для исходной последовательности.
Рис. 1. Выдача Blast для исходной последовательности.
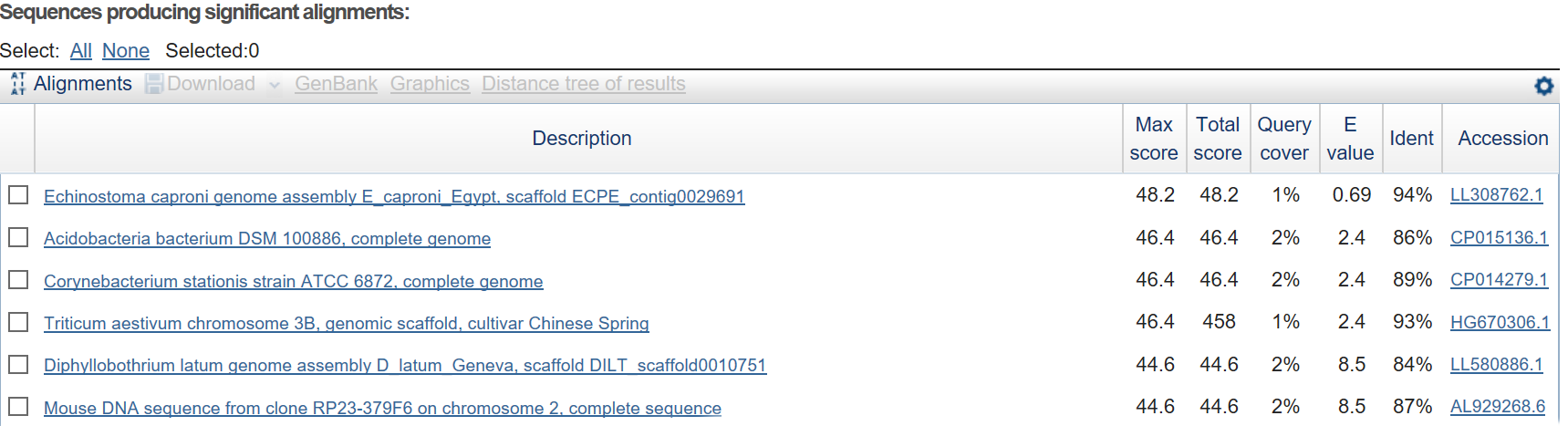 Рис. 2. Выдача Blast для перемешанной последовательности.
Рис. 2. Выдача Blast для перемешанной последовательности.
Как видно на рисунке 2, для перемешанной последовательности было найдено всего 6 последовательностей,
которые имели e-value больше 0.6, query cover очень низкий(2% и менее).
Для исходной последовательности, было найдено 4 находки с низким e-value(ниже 0.01) и 1 находка с очень хорошим E-value и query cover=100%(приведены на рисунке 1), что подтверждает тот факт,
что этот параметр позволяет отсеивать недостоверные находки.
11. С помощью программы cusp были найдены частоты кодирующих кодонов файла h.fasta .
Для этого использовалась команда:
cusp h.fasta
Результат в файле hq608513.cusp.
12. С помощью compseq найдены частоты динуклеотидов в данной нуклеотидной последовательности b.fasta
и сравнены их с ожидаемыми. Использовалась программа compseq и опции -word 2 и -calcfreq.
Для этого использовалась команда:
compseq b.fasta b.compseq -word 2 -calcfreq
В выходном файле b.compseq содержатся частоты динуклеотидов и их отношение к соответствующим ожидаемым частотам.
13. С помощью программы tranalign выровнены кодирующие последовательности a.fasta и c.fasta
соответственно выравниванию белков - их продуктов.
Tranalign принимает на вход набор из невыровненных нуклеотидных последовательностей и соответствующий им набор выровненных транслированных белковых
последовательностей. В выходной файл записывается выравнивание нуклеотидных последовательностей. Каждая нуклеотидная последовательность
транслируется во всех трех прямых рамках по указанному генетическому коду и трансляции сравниваются с соответсвующими во входном выравнивании.
Важно, чтобы соответствующие друг друг последовательности во входных файлах располагались в одном порядке.
Для работы в качестве входных файлов были взяты следующие файлы: n.fasta - невыровненные нуклеотидные последовательности, p.fasta
- выровненные белковые последовательности.
При этом использовалась команда:
tranalign n.fasta p.fasta align.fasta
Результат в файле align.fasta.
Задание 2a.
Для выполнения задания №2 были взяты различные штаммы бактерии Achromobacter xylosoxidans: A8, C54, NH44784-1996
с индентификаторами embl CP002287, CP009448 и HE798385.1 соответственно.
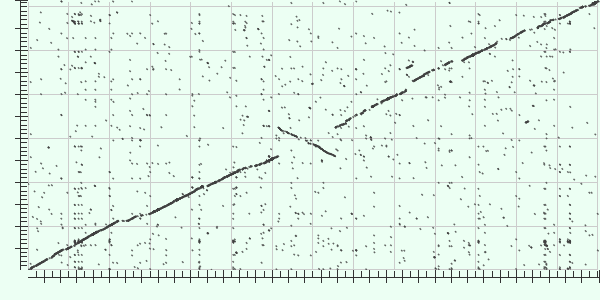
Рисунок 2.Карта локального сходства.
Как видно из рисунка произошла инверсия и 5 вставок(инсерций). Это вставки небольших участков, намного меньших, чем тот, который был подвержен инверсии. Query cover составляет 74%.
Сходство(Identity) между гомологичными участками составляет 86%.