Часть I: подготовка чтений.
fastqc chr21.fastqВывод программы:
Started analysis of chipseq_chunk4.fastq Approx 10% complete for chipseq_chunk4.fastq Approx 25% complete for chipseq_chunk4.fastq Approx 40% complete for chipseq_chunk4.fastq Approx 55% complete for chipseq_chunk4.fastq Approx 65% complete for chipseq_chunk4.fastq Approx 80% complete for chipseq_chunk4.fastq Approx 95% complete for chipseq_chunk4.fastq Analysis complete for chipseq_chunk4.fastqВ результате были получены 2 файла:chr21_fastqc.zip и chr21_fastqc.html.
Результаты в формате .html: chipseq_chunk4_fastqc.html.
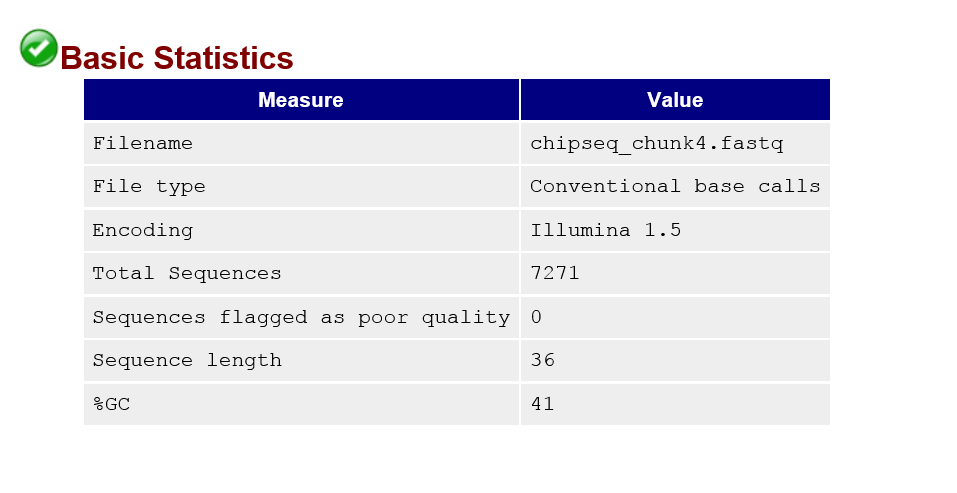
Рис. 1. Информация о количестве ридов, откартированных на геном.
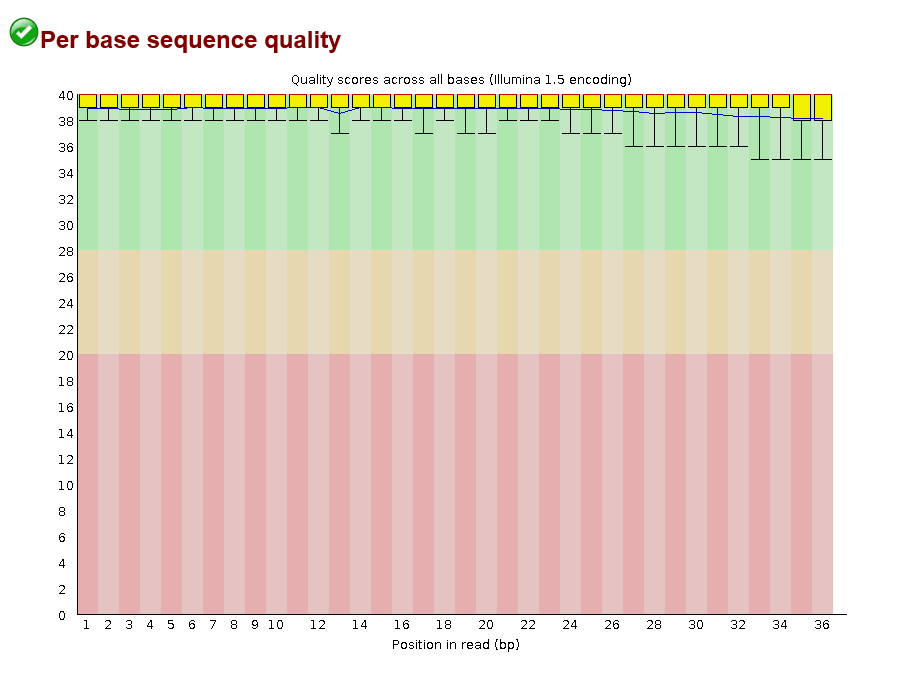
Рис. 2. Информация о количестве ридов, откартированных на геном.
Очистка чтений программой Trimmonatic не требуется.
Далее последовательности были откартированы на проиндескированный геном человека.
Использованная команда:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk4.fastq > chipseq_chunk4.samВыдача:
[M::main_mem] read 7271 sequences (261756 bp)... [M::mem_process_seqs] Processed 7271 reads in 1.996 CPU sec, 2.025 real sec [main] Version: 0.7.10-r789 [main] CMD: bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk4.fastq [main] Real time: 61.135 sec; CPU: 15.036 secПосле этого был проведен анализ полученного файла с помошью следующих команд:
Перевод в бинарный формат: samtools view -b chipseq_chunk4.sam -o chunk4.bam
Сортировка по координате в референсе начала чтения: samtools sort chunk4.bam -T chip_temp -o chipseq_chunk4.sorted.bam
Индексация: index chipseq_chunk4.sorted.bam
Получение информации о количестве откартированных ридов: samtools idxstats chipseq_chunk4.sorted.bam > chipseq_chunk4.idxstats
Просмотр количества откартированных ридов: samtools view -c chipseq_chunk4.sorted.bam
Получение количества откартированных ридов:samtools idxstats chipseq_chunk4.sorted.bam
Откартировался 7271 рид. 7235 ридов откартировалось на геном.
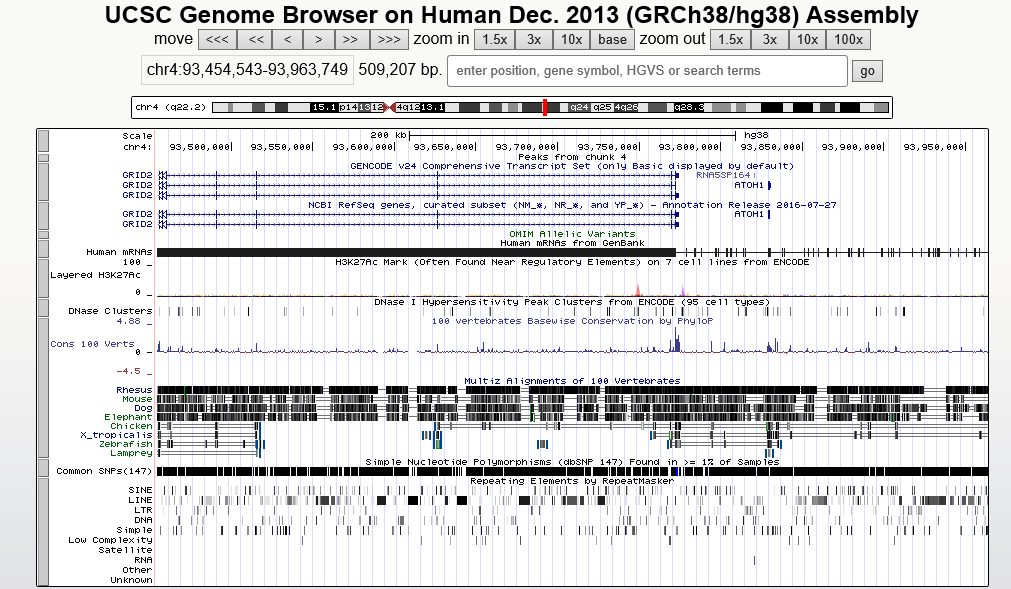
Подавляющее большинство ридов откартировались на chr12. Это позволяет предположить, что для анализа были предложены прочтения с 12 хромосомы. Полная информация о количестве откартированных ридов представлена в файле. Информация о количестве ридов, откартированных на геном представлена на Рисунке 1.
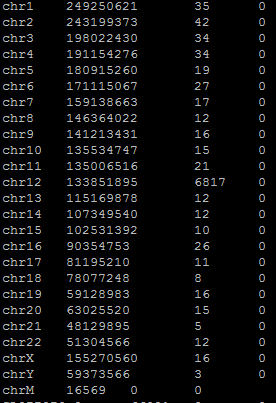
Рис. 1. Информация о количестве ридов, откартированных на геном.
Для поиска пиков была использована программа MACS. Попытка запуска командой: macs2 callpeak -t chipseq_chunk4.sorted.bam не дала результата, так как программа выдала 0 пиков. Далее я запустил программу со следущими параметрами: macs2 callpeak -n chunk4 -t chunk4_sorted.bam --nomodel.
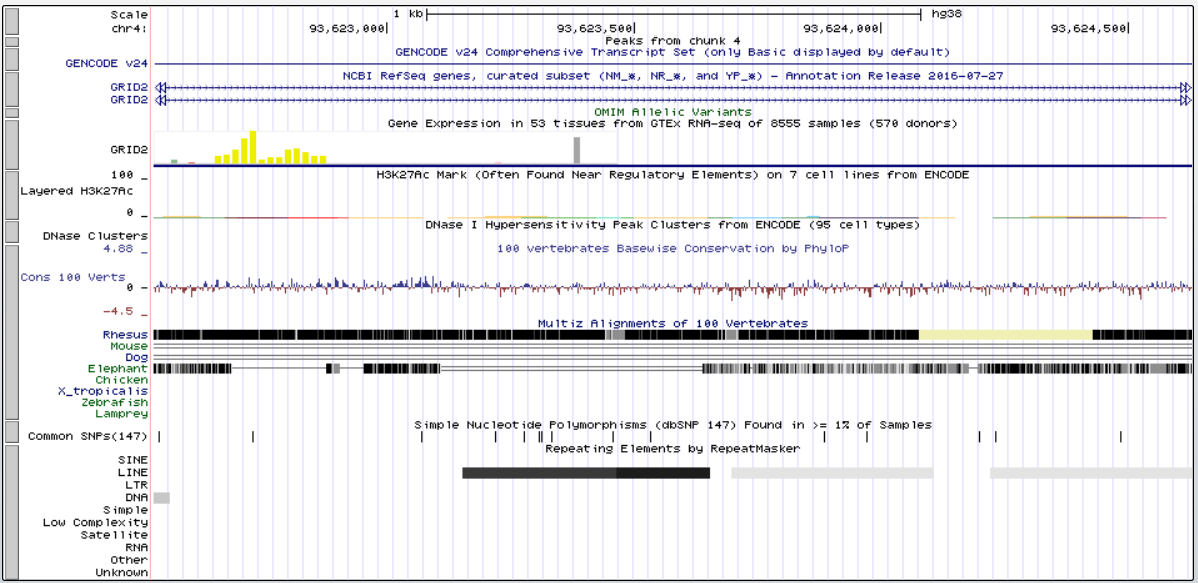
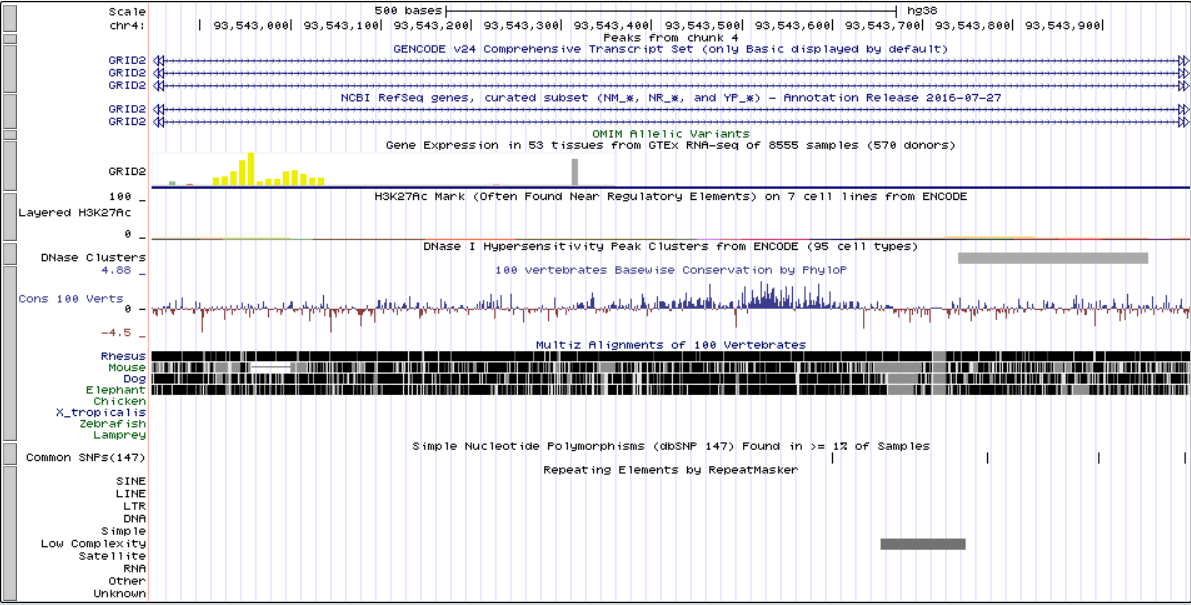
Программа выдала следующие файлы:NA_peaks.narrowPeak , NA_peaks.xls, NA_summits.bed. Наиболее полная информация представлена в файле NA_peaks.xls. Программа нашла 8 пиков. Все пики находятся одном регионе 5 хромосомы.
В Таблице 1 представлена информация о пиках.