1. Таксономия и функция нуклеотидной последовательности
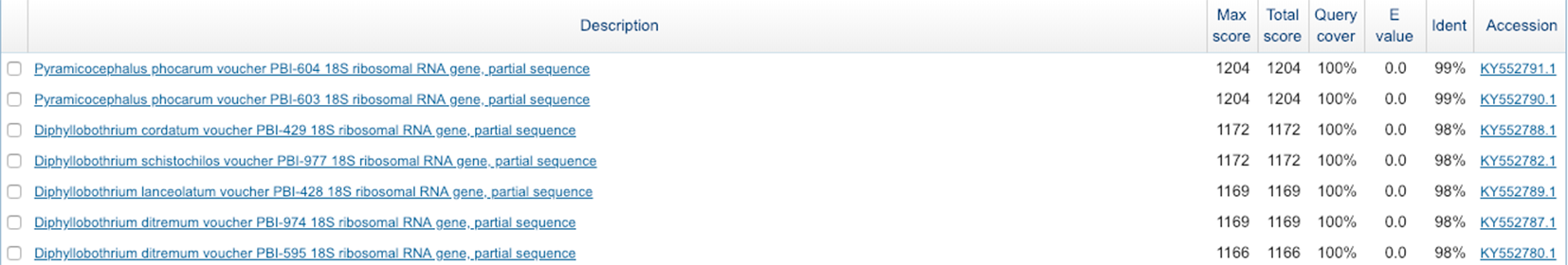
Рисунок 1. Часть выдачи BlastN
Две лучшие находки с Ident 99% относятся к роду Pyramicocephalus. Помимо них есть еще 28 находок с чуть меньшим Ident 98%, относящихся к другому роду - Diphyllobothrium. Исходя из дерева, построенного BLAST по находкам, можно предположить принадлежность именно к первому роду, Pyramicocephalus. Дерево дает представление об эволюционном расстоянии между различными последовательностями, измеряемом количеством мутаций, приводящих к отдалению этих последовательностей друг от друга. Можно видеть, что наша последовательность находится в пределах одной клады, объединяющей представителей вида Pyramicocephalus phocarum - паразита беломорской трески.
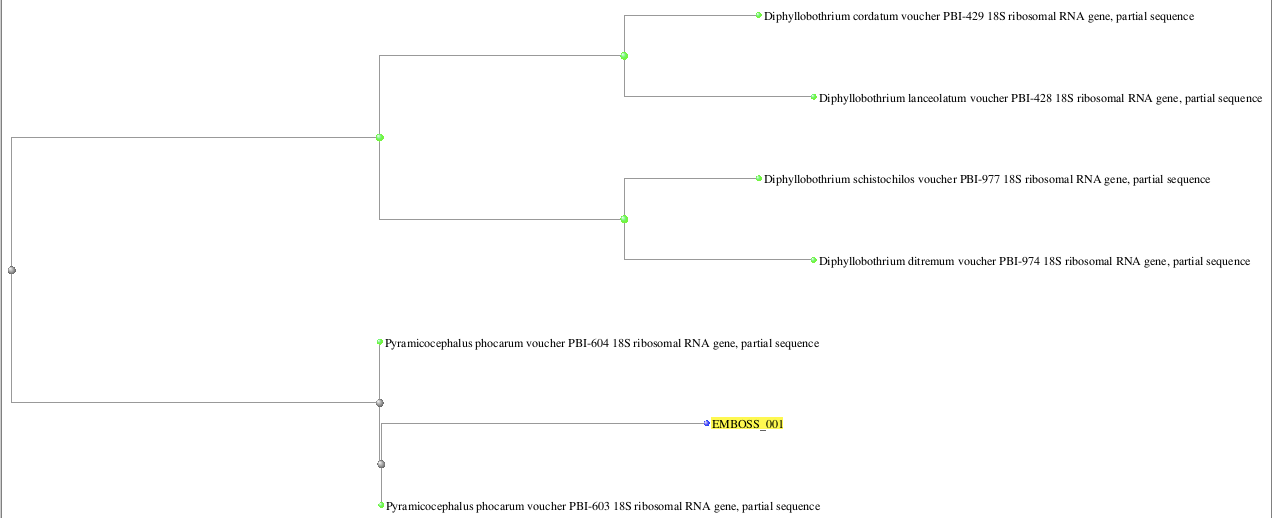
Рисунок 2. Эволюционное дерево находок
2. Сравнение разных вариантов blast
Первый запуск
Для начала сравним список находок трех разных вариантов BLAST для последовательности из первого задания. Гомологов будем искать по банку Nucleotide collection, классу Сestoda, исключив семейство Diphyllobothriidae, к которому принадлежит Pyramicocephalus phocarum. Максимальное количество находок для всех трех вариантов увеличиваем до 1000. Также были поставлены галочки в полях Models (XM/XP) и Uncultured/environmental sample sequences.
| Word size | Match/mismatch | Hits | |
| Megablast | 28 | 1/-2 | 970 |
| BLASTN стандартный | 11 | 2/-3 | 980 |
| BLASTN чувствительный | 7 | 1/-1 | 975 |
Запуск Megablast с описанными выше параметрами дал 970 находок. Все выравнивания хорошие, E-value каждой не привышает 0.0, Ident находится в пределах 84-98%. Вероятно, это обусловлено высокой консервативностью последовательности у разных видов.
BLASTN со стандартными параметрами добавил к выдаче Megablast всего десять находок, смысла в которых немного, так как все они имеют очень низкий coverage. Результат работы чувствительного BLASTN несильно отличается от результата работы стандартного.

Рисунок 3. BLASTN со стандартными настройками

Рисунок 4. BLASTN со чувствительными настройками
Второй запуск
Во втором запуске проводился поиск гомологов последовательности rRNA большой субЪединицы рибосомы из митохондриального генома Bombus hypocrita sapporensis. Поиск проводился по всем артроподам. Даже после исключения целого отряд перепончатокрылых, для которых все алгоритмы давали одинаковые результаты с высоким весом и Identity score из-за высокой консервативности участка, кодирующего рРНК, количество находок для всех видов BLAST превышало порог в 2000.
| Word size | Match/mismatch | Hits | |
| Megablast | 28 | 1/-2 | 2035 |
| BLASTN стандартный | 11 | 2/-3 | >5000 |
| BLASTN чувствительный | 7 | 1/-1 | 975 |
3. Гомологичные белки
Задание было выполнено на трех белках: PRPC_EMENI, TBB_NEUCR и HSP71_YEAST. Так как последовательности белковые, необходимо использовать tblastn. Например, для PRPC_EMENI команда будет выглядеть так:
$ tblastn -query PRPC_EMENI.fasta -db X5.fasta > PRPC_EMENI.out
Пример вывода программы для того же белка:
Score E
Sequences producing significant alignments: (Bits) Value
scaffold-693 393 6e-121
scaffold-157 390 6e-120
scaffold-287 64.3 9e-11
scaffold-212 57.4 1e-08
PRPC_EMENI кодирует белок mcsA. Это митохондриальный ген. Катализирует синтез (2S,3S)-2-метилцитрата из пропионил-КоА и оксалоацетата, а также из ацетил-КоА и оксалоацетата с большей эффективностью. Также обладает активностью цитрат-синтазы. Лучшее выравнивание - со scaffold-693. В целом, E-value достаточно низок, чтобы утверждать гомологию двух белков, но вес недостаточно велик, чтобы однозначно утверждать об одинаковой функции.
TBB_NEUCR
Кодируемый белок - tub-2. Бета-цепи тубулина - важнейшего компонента микротрубочек. Лучшая находка: unplaced-665 (Score: 742, E-val: 0.0). Есть длинные идентичные участки без гэпов. Чистая гомология с длинными участками без гэпов.
HSP71_YEAST
HSP71_YEAST кодирует Heat shock protein SSA1 у Saccharomyces cerevisiae (strain ATCC 204508 / S288c). Белок выполняет функцию транспорта полипептидов через митохондриальную мембрану и в ЭПР. Выравнивание со scaffold-199 обладает самым высоким весом и E-value = 0.0. На этом основании две последовательности можно назвать гомологичными и они, скорее всего, выполняют одну и ту же функцию.
4. Поиск гена в контиге
Для поиска был выбран большой контиг unplaced-1071. Поиск проводился с помощью blastx по базе данных Reference proteins. В контиге длиной 86429 пар оснований был найден ген, кодирующий АТФ-зависимую ДНК лигазу 1.
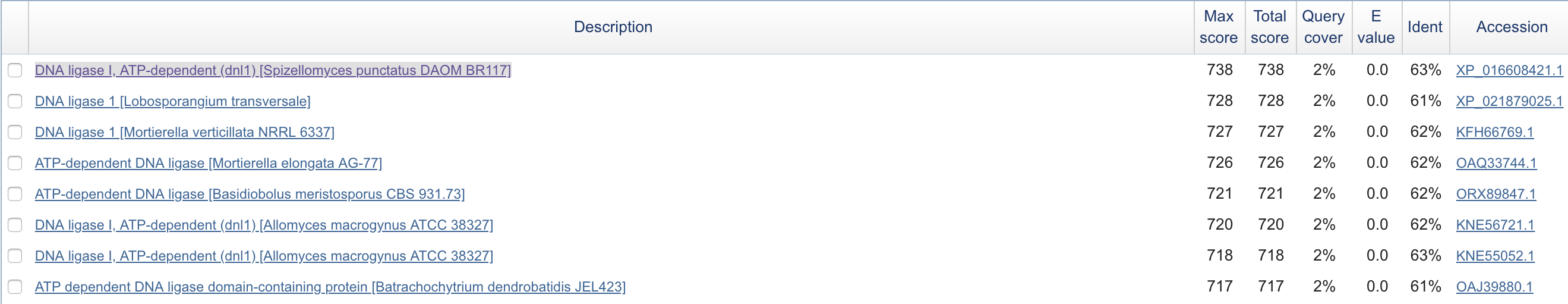
Низкий E-value вызван большой длиной контига и короткой длиной интрона.
5. Карта локального сходства геномов двух бактерий
Для сравнения были выбраны двуе бактерии рода Brucella: Brucella ovis (NC_009504.1) и Brucella abortus (NC_006933.1).
Ниже изображена карта локального сходства геномов этих бактерий. На вертикальная оси расположена последовательность B.ovis, на горизональной - B. abortus. Можно видеть, разрыв в правом вехнем углу свидетельствует о том, что у B. ovis произошла делеция, в центре четко видна инверсия.
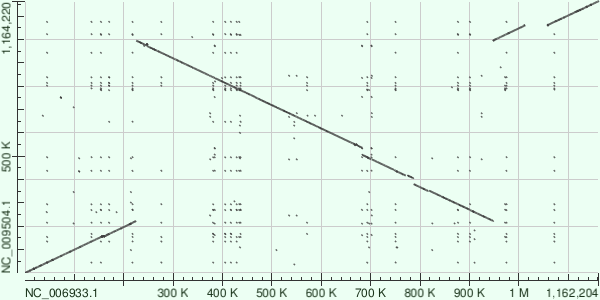
Рисунок 5. Карта локального сходства геномов B.ovis и B. abortus.
BlastN был запущен для консенсусной последовательности из вырвнивания прямой и обратной последовательностей, полученном в практикуме 6. Все записи соответствуют гену 18S rRNA, поэтому принадлежность нашей последовательности именно к этому типу генов не вызывает сомнений.