
Рисунок 1. До чистки
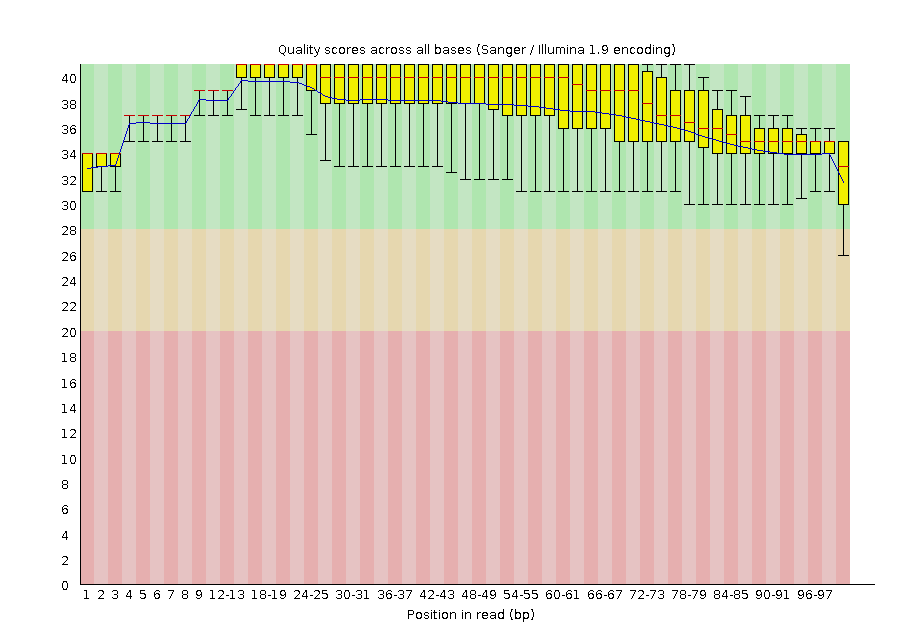
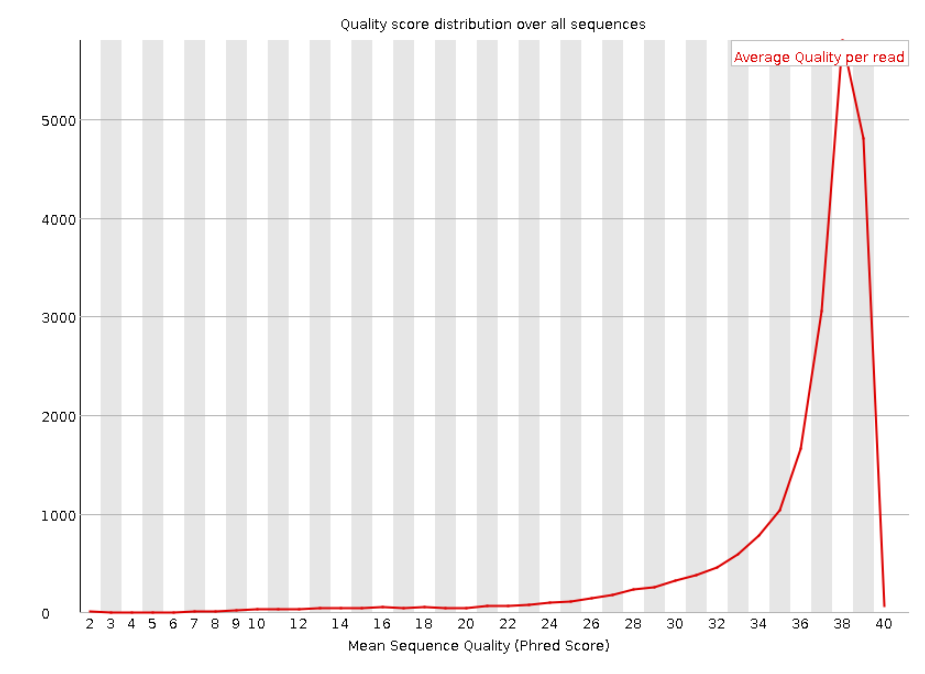
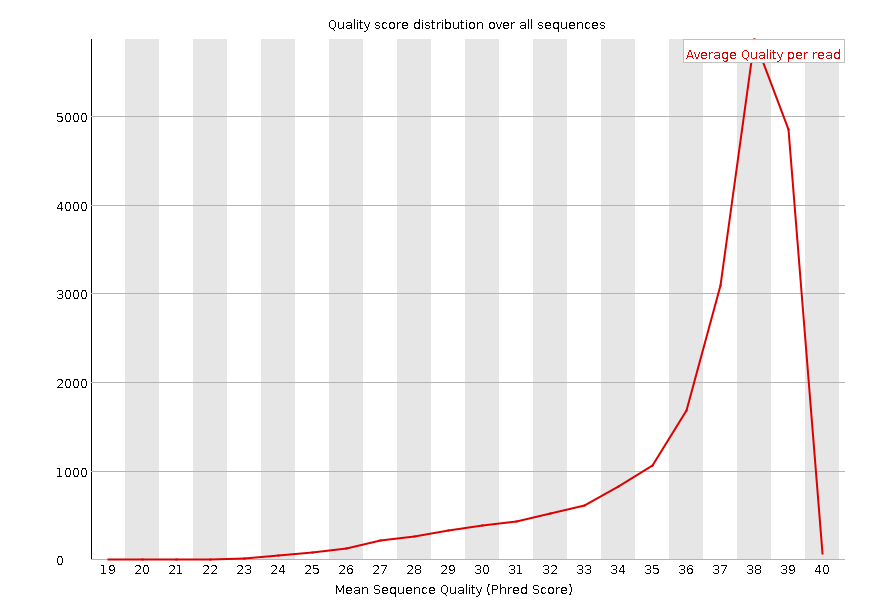
| Тип | Координата | В референсе | В прочтении | Качество прочтения | Глубина прочтения |
| Замена | 41291081 | G | A | 221.999 | 25 |
| Делеция | 41577856 | gacaaaca | gaca | 22.4955 | 1 |
| Вставка | 41824696 | G | GT | 4.4191 | 1 |
| 1 | Анализ качества чтений | Что сделанно |
|---|---|---|
| fastqc chr3.fastq | проведен контроль качества с помощью программы FastQC | |
| 2 | Очистка чтений | |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq chr3_new2.fastq TRAILING:20 MINLEN:50 | проведена очистка чтений с помощью программы Trimmomatic | |
| 3 | Картирование чтений | |
| hisat2-build chr3.fasta chr3_his.fasta | проиндексирована референсная последовательность | |
| hisat2 -x chr3_ршы.fasta -U chr3_new2.fastq --no-spliced-alignment --no-softclip > 1.sam | построиенно выравнивание прочтений и референса в формате "*.sam" | |
| 4 | Анализ выравнивания | |
| samtools view 1.sam -b -o 1.bam | переведено выравнивание чтений с референсом в бинарный формат "*.bam* | |
| samtools sort 1.bam -T 0.txt -o sort.bam | отсортировано выравнивание чтений с референсом по координате в референсе начала чтения | |
| samtools index sort.bam | проиндексирован отсортированный "*.bam" файл | |
| samtools idxstats sort.bam > task4.txt | записано числа откартировавшихся чтений | |
| 5 | Поиск SNP и инделей | |
| samtools mpileup -uf chr3.fasta sort.bam > 1.bcf | создан файл с полиморфизмами в формате "*.bcf* | |
| bcftools call -cv 1.bcf > 1.vcf | создан файл со списком отличий между референсом и чтениями в формате "*.vcf* | |
| 6 | Аннотация SNP | |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 1.vcf > 1.avinput | конвертирован файл из "*.vcf* в "*.avinput" | |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1 -build hg19 -dbtype 1138 1.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных dbsnp | |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1000g -buildver hg19 -dbtype 1000g2014oct_all 1.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных 1000 genomes | |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.gwas -build hg19 -dbtype gwasCatalog 1.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных Gwas | |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.clinvar -dbtype clinvar_20150629 -buildver hg19 1.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных Clinvar | |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out rs.refgene -build hg19 1.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных refgene |
© Угольков Ярослав, 2017