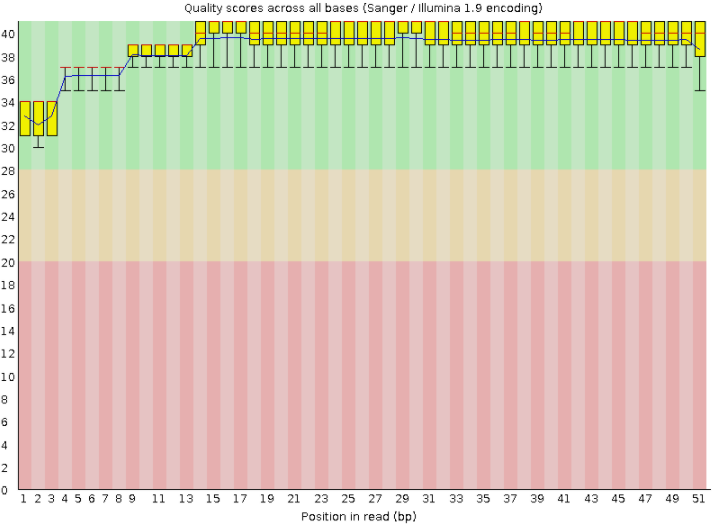
Рисунок 1.
| 1 | Анализ качества чтений | Что сделанно |
|---|---|---|
| fastqc chr3.1.fastq | проведен контроль качества с помощью программы FastQC | |
| 2 | Картирование чтений | |
| hisat2-build chr3.fasta chr3_old.fasta | проиндексирована референсная последовательность | |
| hisat2 -x chr3_old.fasta -U chr3.1.fastq -S qw.sam | построиенно выравнивание прочтений и референса в формате "*.sam" | |
| 3 | Анализ выравнивания | |
| samtools view qw.sam -b -o qw.bam | переведено выравнивание чтений с референсом в бинарный формат "*.bam* | |
| samtools sort qw.bam -T 1.txt -o sort1.bam | отсортировано выравнивание чтений с референсом по координате в референсе начала чтения | |
| samtools index sort1.bam | проиндексирован отсортированный "*.bam" файл | |
| samtools idxstats sort1.bam > task.txt | записано числа откартировавшихся чтений | |
| 4 | Подсчет чтений | |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtobed -i qw.bam > qw.bed | cоздан файла в формате "*.bed" из файла формата "*.bam" | |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools intersect -a | найдено пересечение выравниваний с разметкой по генам | |
| /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b qw.bed -c > er.bed | ||
| sort -k 6 -r er.bed > sort_er.bed | создан отсортированный файл |
[1] Wikipedia, TFRC, 2015
© Угольков Ярослав, 2017