Ресеквенирование. Поиск полиморфизмов у человека
Мне была дана хромосома 16 и, соответственно файлы chr16.fasta и chr16.fastq.
1. Анализ качества чтений
С помощью программы FastQC был осуществлен контроль качества чтений. В результате работы команды fastqc chr16.fastq получена html страница chr16_fastqc.html.
2. Очистка чтений
Далее была сдеалана очистка чтений с помощью программы Trimmomatic. Команда
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr16.fastq trimm_out.fastq MINLEN:50 TRAILING:20
удаляет прочтения короче 50 (MINLEN:50) и
удаляет нуклеотиды ниже качества "20" с конца прочтений (TRAILING:20). В результате получен файл trimm_out.fastq.
Необходимо провести анализ прочтений после чистки и сравнить с результатом до него.
Командой fastqc trimm_out.fastq получена страница trimm_out_fastqc.html
До очистки было 3965 прочтений, после - 3962.
|
|
|
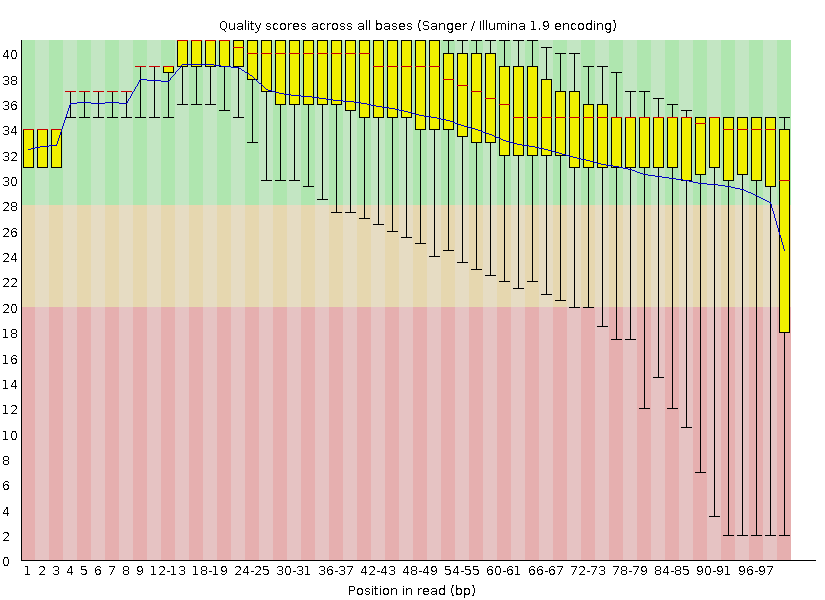
Рис. 1 Per base sequence quality до очистки |
|
|
|
Рис. 2 Per base sequence quality после очистки |
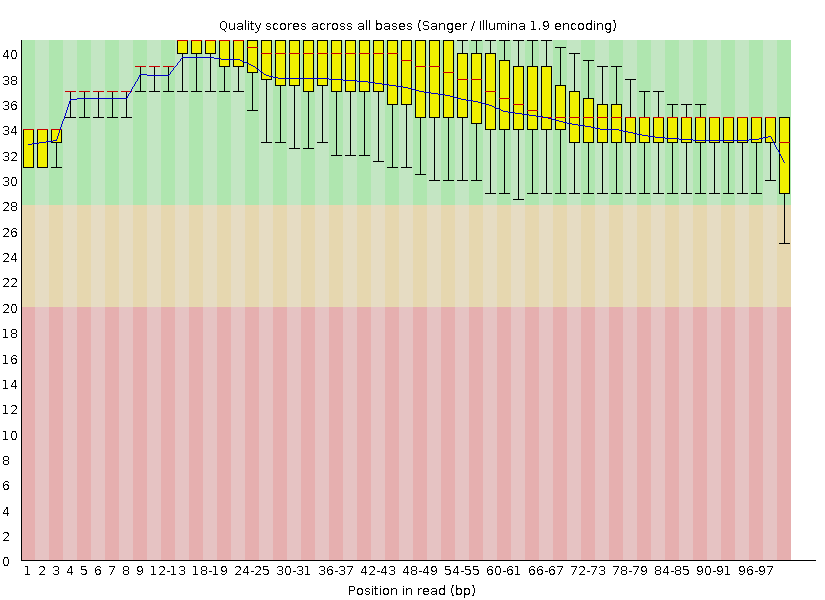
На рисунках 1 и 2 показаны качества прочтений до и после очистки по всем основаним в каждой позиции FastQ файла.
Для каждой позиции желтый прямойгольник - интерквартильный размах (от 25 о 75%),концы черных линий - точки от 10 до 90%, красная линия - медиана,
синяя линия - математическое ожидание.
Чем выше значение по оси Y, тем лучше определено основание. Области фона также говорят о точности: зеленая - качество хорошее, оранжевая - среднее, красная - плохое.
Видно, что до очистки качество было значительно хуже, в то время, как после все чтения оказались в "зеленой области".
3. Картирование чтений
Далее неоходимо провести картирование чтений с помощью программы BWA.
Командой bwa index chr16.fasta хромосома 16 была проиндексирована.
Затем, команой bwa mem chr16.fasta trimm_out.fastq >> aiign_pr13.sam было построено выравнивание прочтений и проиндексированной референсной последовательности (align_pr13.sam).
4. Анализ выравнивания
Выравнивание было преведено в бинарный формат bam команой
samtool view -b align_pr13.sam -o align_pr13.bam
Параметр -b обозначает формат выходного файла (bam), -o - выходной файл (align_pr13.bam).
Далее выравнивания чтений с референсом были отсортированы по координате начала чтения в референсе командой
samtools sort align_pr13.bam align_sort (выходной файл align_sort.bam)
Командой samtools index align_sort.bam отсортированный файл был проиндексирован.
Было выяснено, сколько чтений откартировалось на геном
samtools idxstats align_sort.bam >> number.out
Откартировались все 3674 прочтения, что остались после очистки.
5. Поиск SNP и инделей
Команой samtools mpileup -uf chr16.fasta align_sort.bam -o snp.bcf был создан bcf-файл с полиморфизмами.
Затем, был создан vcf-файл со списком отличий между референсом и чтениями командой bcftools call -cv snp.bcf -o snp.vcf.
Всего получено 62 SNP и 2 инделя.
В таблице №1 описаны 3 полиморфизма из файла snp.vcf.
| Таблица №1 Информация о полиморфизмах |
|||||
| Позиция | В референсе | В чтениях | Тип полиморфизма | Глубина покрытия | Качество чтений |
| 11444454 | gaaaaaaaaaaa | gaaaaaaaaaa | делеция | 74 | 4,40924 |
| 11362640 | G | A | замена | 48 | 166.009 |
| 11348246 | ACACTC | ACACTCACTC | вставка | 5 | 10,8393 |
6. Аннотация SNP
С помощью программы annovar было необходимо проаннотировать только полученные snp, для чего из файла snp.vcf вручную были удачены индели (snp_only.vcf).
Чтобы получить из файла snp_only.vcf получить файл, с которым может работать программа annovar,
была использована команда
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_only.vcf -outfile snp.avinput.
Затем, полученный файл snp_only.avinput использовался для аннотации SNP по базам данных refgene, dbsnp, 1000 genomes, GWAS и Clinvar с помощью скрипта annotate_variation.pl.
1. Аннотация по базе refgene
После работы команды
perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr16.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
были получены файлы chr16.refgene.variant_function (описание всех SNP), chr16.refgene.exonic_variant_function (описание SNP, попавших в экзоны) и chr17.refgene.log (описание процесса работы команды).
В таблице №2 указана информация о SNP в различных группых, полученная из файла chr16.refgene.variant_function.
В таблице №3 представлена информация о SNP в экзонах, полученная из файла chr16.refgene.exonic_variant_function. Всего в экзоны попало 9 SNP.
|
Таблица №2 Данные о SNP в различных группых |
||||
| В интронах | В экзонах | UTR3 | Гомо | Гетеро |
| 22 | 9 | 3 | 37 | 25 |
|
Таблица №3 Данные о SNP в экзонах |
||||||||
| Координата | Тип замены | Ген | В референсе | В чтениях | Замена аминокислоты | Гетеро или гомозиготная замена | Качество чтений | Глубина покрытия |
| 11362729 | несинонимичная | TNP2 | G | A | R131W | гетерозиготная | 225,009 | 144 |
| 11367143 | stopgain | PRM3 | G | A | R104X | гомозиготная | 221,999 | 32 |
| 11367154 | несинонимичная | PRM3 | C | T | R100Q | гомозиготная | 222 | 27 |
| 11369938 | несинонимичная | PRM2 | G | A | P97L | гетерозиготная | 142,008 | 24 |
| 11369947 | несинонимичная | PRM2 | G | A | A94V | гетерозиготная | 144,008 | 24 |
| 11374866 | синонимичная | PRM1 | G | T | R47R | гомозиготная | 221,999 | 38 |
| 11444572 | синонимичная | RMI2 | A | C | T123T | гетерозиготная | 225,009 | 110 |
| 31096164 | несинонимичная | PRSS53 | G | C | P406A | гетерозиготная | 148,077 | 7 |
| 56969148 | несинонимичная | HERPUD1 | G | A | R50H | гетерозиготная | 225,009 | 79 |
Как видно из таблиц, большинство замен произошли в интронах. Это можно объяснить тем, что замены в кодирующих последовательностях могут привести в изменению аминокислоты в конечном продукте (белке), тем самым нарушив его функцию.
2. Аннотация по базе dbsnp
В результате работы команды
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr16.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
были получены файлы chr16.dbsnp.hg19_snp138_dropped (SNP с идентификатором rs), chr16.dbsnp.hg19_snp138_filtered (SNP без rs) и chr16.dbsnp.log (описание работы программы).
56 SNP имеют аннотацию в базе данных dbSNP, а 6 не имеют.
3. Аннотация по базе 1000 genomes
Командой /nfs/srv/databases/ngs/ulyana$ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr16.1000 -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
были получены 3 файла chr16.1000.hg19_ALL.sites.2014_10_dropped, chr16.1000.hg19_ALL.sites.2014_10_filtered, chr16.1000.log, аналогичные полученным в предыдущем пункте.
56 имеют аннотация, а 6 не имеют.
Также мы смогли узнать частоты аннотированных полиморфизмов. Она варьируется от 0,004992 до 0,984026. Среднее значение равно 0,577354.
4. Аннотация по базе GWAS
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr16.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
Получено 2 файла chr16.gwas.hg19_gwasCatalog ((SNP, для которых известно клиническое значение) и chr16.gwas.log.
Было найдено 3 таких SNP. Один связан с ожирением, другой вызывает предрасположенность к болезни Паркинсона, третий - к метаболическому синдрому.
5. Аннотация по базе Clinvar
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr16.clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput
/nfs/srv/databases/annovar/humandb/
Получено 3 файла chr16.clincar.hg19_clinvar_20150629_dropped, chr16.clincar.hg19_clinvar_20150629_filtered, chr16.clincar.log.
Оказалось, что в этой базе данных ни один из изучаемых SNP не аннотирован.
Итоговый файл по всем SNP: pr13.xlsx.