EMBOSS: пакет программ для анализа последовательностей
Задание 1. Отчет о выполнении упражнений
1. Несколько файлов в формате fasta собрать в единый файл
В файле list.txt находится список последовательностей в формате usa. Команда
seqret @list.txt sequences.fasta
записывает в файл sequences.fasta сами последовательности.
2. Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
С помощью команды
seqretsplit sequence.fasta
из файла sequences.fasta были получены файлы p53_human.fasta,
egfr_human.fasta, src_human.fasta.
3. Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
В файле list2.txt записаны данные о нужных участках (координаты "от", "до", "ориентация"). Команда
seqret @list2.txt seq2.fasta
записывает нужные участки из файла chr.gb в файл seq2.fasta.
4. Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле
Команда
transeq seq2.fasta prot.fasta
транслирует нуклеотидные последовательности из файла seq2.fasta в последовательнотсь аминокислот в файле
prot.fasta.
5.Транслировать данную нуклеотидную последовательность в шести рамках
Команда
transeq chr.gb 6frames.fasta -frame 6
транслирует в шести рамках последовательность из файла chr.gb в файл
6frames.fasta.
6. Перевести выравнивание и из fasta формате в формат .msf
Команда
seqret align.fasta msf::align.msf
сохраняет файл align.fasta в формате msf.
7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число)
Команда
infoalign align.msf -only -name -idcount stdout
выдает в выходной поток следующее:
|
|

8. Перевести аннотации особенностей в записи формата .gb в табличный формат .gff
Команда
chr.gb -auto
переводит файл chr.gb в файл chr.gff.
9. Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями
Команда
extractfeat chr.gb cds.fasta -type cds -describe product
выдает в файле cds.fasta все кодирующие последовательности и описание белка, полученные из файла
chr.gb.
10. Перемешать буквы в данной нуклеотидной последовательности
Команда
shuffleseq lal.fasta kek.fasta
перемешивает последовательность в файле lal.fasta; получаем kek.fasta.
А бласт работать не хочет (хотя я тоже не хочу, хочу спать).
11. Найдите частоты кодонов в данных кодирующих последовательностях
Команда
cusp cds.fasta azaza.table
создает таблицу azaza.table частот кодонов в кодирующих последовательностях файла cds.fasta.
12. Найдите частоты динуклеотидов в данной нуклеотидной последовательности и сравните их с ожидаемыми
С помощью команды
compseq lal.fasta lalka.hz -word 2 -calcfreq
получаем файл lalka.hz - таблицу частот динуклеотидов последовательности lal.fasta.
В файле представлено общее количество каждого из динуклеотидов, реальная, ожидаемая частоты и их отношение.
13. Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов
Команда
tranalign kekmakek.fasta ololo.fasta tranalign.fasta
из набора невыровненных нуклеотидных последовательностей kekmakek.fasta
и набора выровненных белковых последовательностей ololo.fasta (транслированных с нуклеотидных) получает выровненные нуклеотидные последовательности tranalign.fasta.
Задание 2a. Построение карты локального сходства и описание крупных эволюционных событий на пути от общего предка
Для построения карты сходства были выбраны Streptococcus suis штамм BM407 (ссылка на геном в fasta формате)
и Streptococcus suis штамм JS14 (ссылка на геном в fasta формате). Геномы Streptococcus suis состоят из одной кольцевой хромосомы.
С момощью алгоритма blast2seq на сайте NCBI была получена карта локального сходства. Для полученного выравнивания e-value - 0.0, identity - 99%, query cover - 96%.
На рисунке 1 представлена полученная карта сравнения геномов. По оси X в ней располагается геном штамма BM407, а по оси Y - штамма JS14. Будем называть их геномы X и Y соответственно.
|
|
| Рис. 1 Карта локального сходства исследуемых последовательностей |
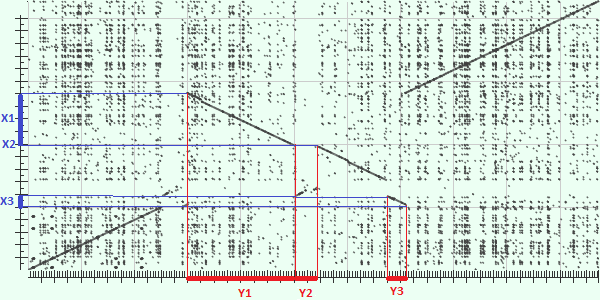
Я выбрала 3 примера крупных эволюционных событий.
1. Участок 1 в геномe X повернут на 180 градусов, относительно генома Y. То есть в одном из геномов произовша инверсия данного участка.
2. Участок 2 отсутствует в геноме X (обозначен точкой X1), что говорит либо о делеции в геноме X, либо о вставле в геноме Y.
3. Участок 3 располагается в геноме X раньше, чем в геноме Y, что говорит о транслокации участка в одном из геномов.
Видно, что гомологичные участки занимют большую часть геномов.