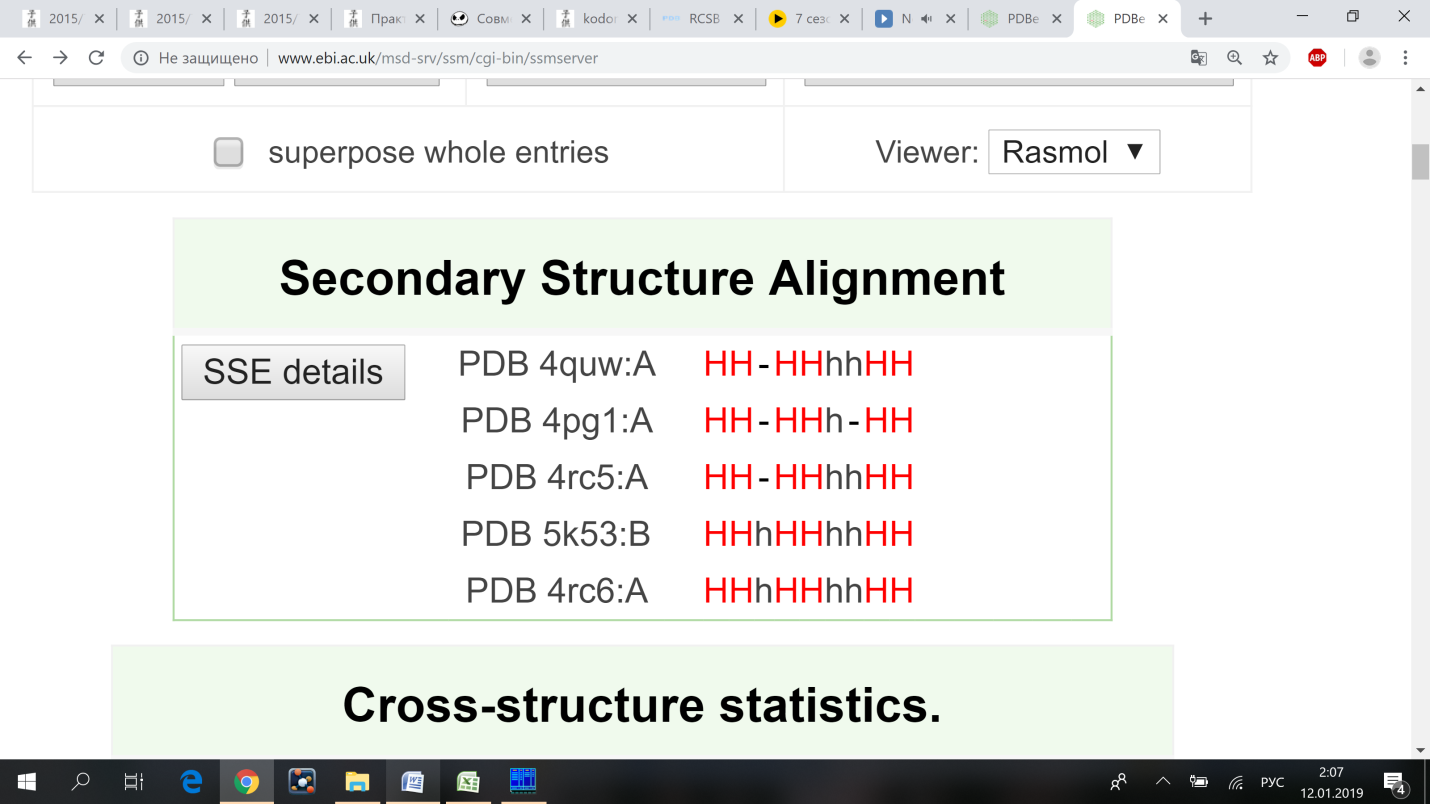
Совмещение структур
Общие характеристики полученного выравнивания следующие: число выровненных а.о. = 215, число выровненных SSE = 6, среднее RMSD = 0.8577, среднее Q-score = 0.8873. Также общая статистика (cross-structure statistics) представлена в таблице №2.
На рис. 1 показано множественное структурное выравнивание выбранных белков с «моим» белком, а на рис. 2 — совмещение структур (из файла < pr8alignment.rasmol>).
|
Таблица №1. Выбранные структуры гомологов и некоторые параметры |
||||||
|
№ |
Q-score |
RMSD |
N_align |
N_align/N_query, % |
PDB ID |
N_SSE |
|
1 |
0.79 |
0.84 |
206 |
0,91964 |
4pg1:A |
7 |
|
2 |
0.89 |
0.86 |
219 |
0,97768 |
4rc5:A |
8 |
|
3 |
0.86 |
0.96 |
216 |
0,96429 |
5k53:B |
9 |
|
4 |
0.84 |
0.98 |
212 |
0,94643 |
4rc6:A |
9 |
|
Таблица №2. Статистика для выбранных структур |
||||||
|
RMSD |
||||||
|
Структура: |
1 |
2 |
3 |
4 |
5 |
|
|
1 |
PDB 4quw:A |
|
1.192 |
1.052 |
0.960 |
0.975 |
|
2 |
PDB 4pg1:A |
1.192 |
|
0.948 |
0.805 |
0.897 |
|
3 |
PDB 4rc5:A |
1.052 |
0.948 |
|
0.484 |
0.454 |
|
4 |
PDB 5k53:B |
0.960 |
0.805 |
0.484 |
|
0.407 |
|
5 |
PDB 4rc6:A |
0.975 |
0.897 |
0.454 |
0.407 |
|
|
Q-score |
||||||
|
Структура:
|
1 |
2 |
3 |
4 |
5 |
|
|
1 |
PDB 4quw:A |
|
0.803 |
0.828 |
0.855 |
0.868 |
|
2 |
PDB 4pg1:A |
0.803 |
|
0.853 |
0.887 |
0.889 |
|
3 |
PDB 4rc5:A |
0.828 |
0.853 |
|
0.927 |
0.947 |
|
4 |
PDB 5k53:B |
0.855 |
0.887 |
0.927 |
|
0.964 |
|
5 |
PDB 4rc6:A |
0.868 |
0.889 |
0.947 |
0.964 |
|
|
Sequence Identity |
||||||
|
Структура: |
1 |
2 |
3 |
4 |
5 |
|
|
1 |
PDB 4quw:A |
|
0.633 |
1.000 |
0.749 |
0.995 |
|
2 |
PDB 4pg1:A |
0.633 |
|
0.633 |
0.628 |
0.628 |
|
3 |
PDB 4rc5:A |
1.000 |
0.633 |
|
0.749 |
0.995 |
|
4 |
PDB 5k53:B |
0.749 |
0.628 |
0.749 |
|
0.744 |
|
5 |
PDB 4rc6:A |
0.995 |
0.628 |
0.995 |
0.744 |
|
Рис. 1. Множественное структурное выравнивание выбранных белков с референсным
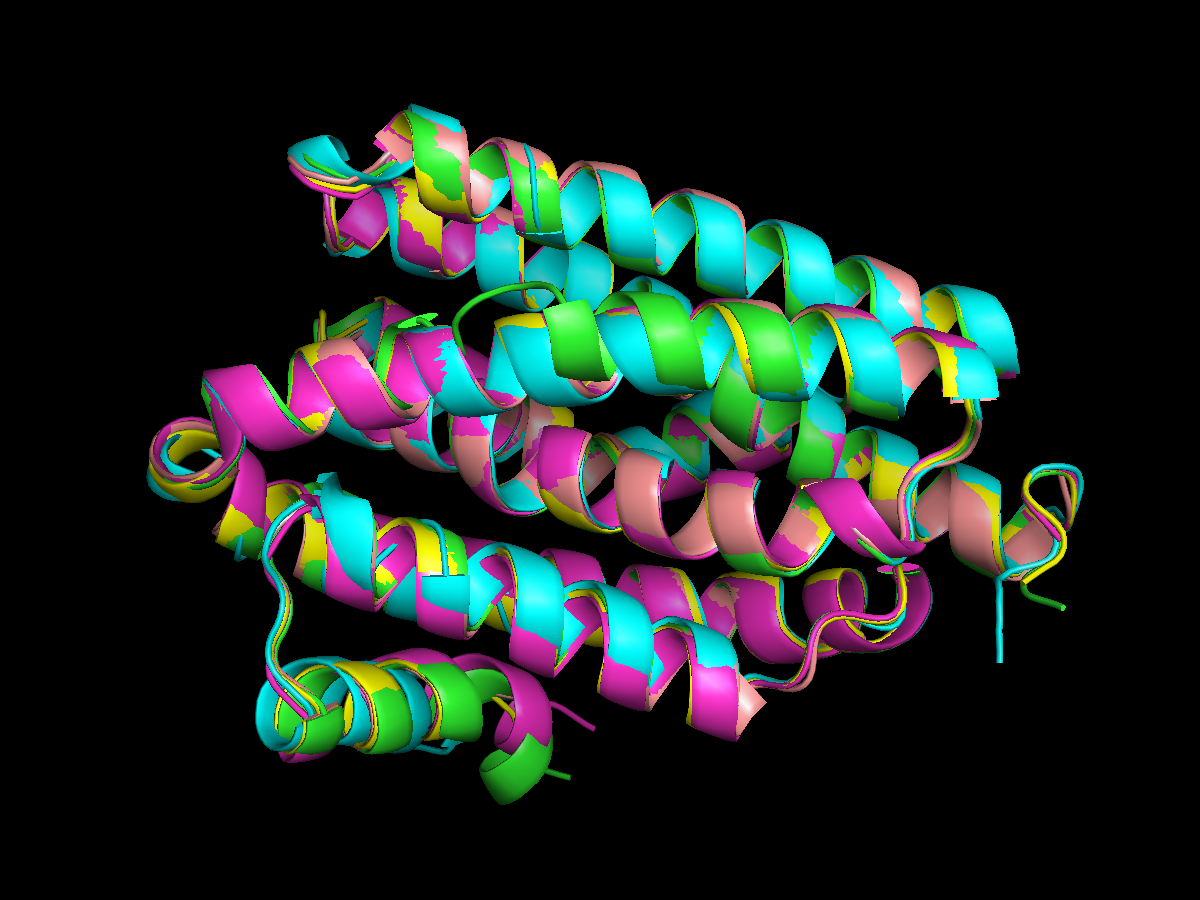
Рис. 2. Совмещение выровненных структур выбранных белков с референсным
Видно, что структуры совмещаются очень хорошо.
Далее были рассмотрены отличия выравнивания последовательностей по структуре (pr8aligned_by_3D.fasta, рис. 3) от выравнивания тех же последовательностей, построенной в программе JalView алгоритмом Muscle (pr8aligned_by_muscle.fasta, рис. 4).
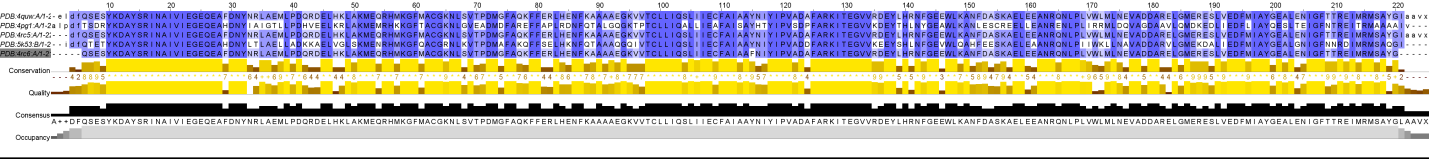
Рис. 3. Выравнивание по структуре
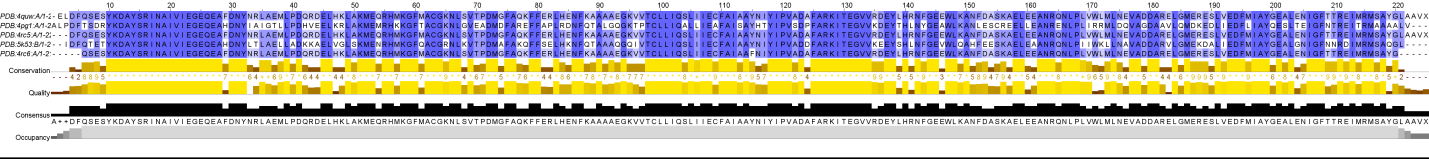
Рис. 4. Выравнивание по последовательностям
Видно, что отличий выравнивания последовательностей нет, кроме небольших не выровненных участков в выравнивании по структуре на концах белков (такие а.о. указаны маленькими буквами). В данном случае, вероятно, это связано с высокой консервативностью вторичной структуры белка.