Консервативный мотив в выравнивании последовательностей гомологичных белков
Для работы было выбрано семейство белковых доменов NurA (PF09376), проявляющих 5'-3'-экзонуклеазную и эндонуклеазную активность и распространённых среди прокариот[1]. Seed-выравнивание содержит 44 последовательности и имеет длину 660 а.о. Файл с выравниванием.
Паттерн мотива: [AGFSTV]-[VITGAL]-D-[GS]-[SGKVH]. Несмотря на большой разброс по буквам, поиск паттерна по выравниванию выдал всего 4 "лишних" находки, помимо 44 верных. Можно заметить, что во всех последовательностях на одной и той же позиции присутствует буква D (аспартат); с учётом её полярности и неполярности других букв из паттерна, этот мотив вполне может быть частью активного центра или играть важную структурную роль.
При работе с сервисом MyHits возникла проблема, т.к. собранный паттерн неспецифичный, и из-за этого сервис находит в базе "too many matches". Поэтому я решил попробовать оптимизировать паттерн, оставив самые консервативные аминокислоты. Для паттерна [AG]-[VIT]-D-[G]-[SG] нашлось 2249 совпадений, для паттерна A-V-D-G-S — 160 сопадений. В первой выдаче четыре находки соответствовали NurA, во второй выдаче — только одна.
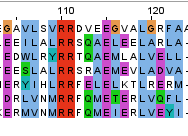
Рис. 1. Выбранный мотив.
Мотив, специфичный для клады филогенетического дерева
Рис. 2. Выбранная клада.
Построение дерева производилось при помощи алгоритма NJ. В дереве я выделил кладу из 7 последовательностей; в их выравнивании можно найти мотив [LIV]..RR[DSTF][QREV].E.{3}L. В общем выравнивании этот мотив не находится больше нигде, кроме этих 7 последовательностей, что говорит о его специфичности.
Рис. 3. Выбранный мотив.
PSI-BLAST
Для работы был выбран белок с идентификатором Q7VDL2 — probable septum site-determining protein MinC, выделенный из бактерии Prochlorococcus marinus. По результатам работы PSI-BLAST составлена таблица:
Идентификатор белка:
Q7VDL2
Номер итерации
Число находок выше порога (0,005)
Идентификатор худшей находки выше порога
E-value этой находки
Идентификатор лучшей находки ниже порога
E-value этой находки
1
146
Q9AG20.1
0.005
A8GFG7.1
0.005
2
188
B6JKX0.1
7.00E-08
A7H8E6.1
0.064
3
188
Q9ZM51.1
2.00E-12
A7H8E6.1
0.014
Количество "хороших" находок перестало изменяться после второй итерации. При этом на второй итерации e-value лучшей из "хороших" и худшей из "плохих" находок различаются на 6 порядков, а на третьей итерации эта разница составляет уже 10 порядков. это говорит о хорошей обособленности данного семейства белков.
Проверка числа ТА в геноме бактерии
В качестве генома была взята последовательность хромосомы Escherichia coli, штамм O157:H7 str. Sakai. Длина составляет 7 890 332 п.о., число букв А и Т равно 1 954 504 (24,8%) и 1 947 037 (24,7%) соответственно. Исходя из этого, ожидаемое число слов ТА равно 482 159,2. Фактическое число таких динуклеотидов равно 365 293 (скрипт), что в 1,32 раза меньше ожидаемого (контраст 0,76).
Для определения достоверности отличия был проведён Z-тест (скрипт). В качестве нулевой гипотезы принято утверждение о том, что среднее число сайтов ТА равно 482 159,2 (ожидаемому). По результатам Z-теста p-value оказалось примерно равным 0. Это говорит о том, что имеющееся отличие имеет статистическую значимость, и нулевая гипотеза может быть отвергнута.
Литература:
Constantinesco F, Forterre P, Elie C; NurA, a novel 5'-3' nuclease gene linked to rad50 and mre11 homologs of thermophilic Archaea, EMBO Rep., 2002.