Предсказание вторичной структуры заданной тРНК и анализ нуклеиново-белкового комплекса
В данном практикуме предлагалось провести предсказание вторичной структуры заданной тРНК путём поиска инвертированных повторов с помощью программы из пакета EMBOSS einverted и по алгоритму Зукера с применением программы ViennaRNA, а также провести поиск ДНК-белковых контактов в заданной структуре.
Предсказание вторичной структуры заданной тРНК путём поиска инвертированных повторов и с помощью алгоритма Зукера
С помощью программы einverted предлагалось воспроизвести вторичную структуру выданной в прошлом практикуме тРНК (PDB ID: 1J1U). Принцип работы этой программы основан на поиске
инвертированных повторов в нуклеотидной последовательности. В случае тРНК программа ищет комплементарные последовательности, которые потенциально могли бы сложиться в шпильки.
Таким образом, была написана следующуя команда:
einverted -sequence tRNA_1J1U.fasta -gap 12 -threshold 10 -match 3 -mismatch -3 -outfile tRNA_1J1U.einvertedгде
-sequnce - параметр, принимающий на вход последовательность РНК в FASTA-формате. В моём случае на вход был передан файл, содержащий последовательность выданной мне тРНК (исходный файл, скачанный со страницы структуры в PDB, содержал также последовательность аминоацил-тРНК-синтетазы, поэтому из итогового файла tRNA_1J1U.fasta она была удалена, однако эмпирическим путём было проверено, что программа и так справляется с поисков комплеменатрных участков);
-gap - штраф за открытие гэпа;
-threshold - минимальный пороговый score, при котором повтор будет считаться значимым;
-match - количество очков, начисляемое за совпадение, т.е. за комплементарную пару нуклеотидов;
-mismatch - штраф на несовпадение, т.е. за некомплементарную пару нуклеотидов;
-outfile - название выходного файла в формате .einverted
В итоге был найден лишь один инвертированный повтор, который, исходя из раннее проведённого анализа структуры тРНК find-pair, представляет собой акцепторный стебель (Файл, содержащий вывод программы einverted). Такой результат можно объяснить тем, что акцепторный стебель является самым протяжённым элементом вторичной структуры тРНК, и, скорее всего, программа посчитала остальные найденные повторы незначимыми.
Далее было предложено провести предсказание вторичной структуры той же тРНК с помощью пакета программ ViennaRNA. Этот пакет программ предназначен для предсказания вторичной структуры молекул
РНК, и в частности, тРНК, с помощью алгоритма Зукера, основанного на поиске структуры с минимальной свободной энергией, используя метод динамического программирования. Предсказание
структуры проводилось с помощью веб-сервиса RNAfold с параметрами по умолчанию. В результате были получены 2 структуры:
вторичная структура тРНК с минимальной свободной энергией и "центроидная" вторичная структура тРНК, отображающее наиболее репрезентативное, усреднённое состояние молекулы из всех возможных
(см. Рис. 1., Рис. 2.). Цветом обозначены вероятности присутствия в структуре тех или иных пар оснований.
.png)
_centroid.png)
Ниже приведена итоговая таблица (см. Таблицу 1) с результатами предсказания вторичной структуры заданной тРНК с помощью программы einverted и веб-сервиса RNAfold, а также сравнение найденных элементов структуры с результатами программы find-pair.
| Элемент вторичной структуры тРНК | Координаты (номера нуклеотидов) элементов вторичной структуры тРНК, определённые программой find-pair | Координаты предсказанных элементов вторичной структуры тРНК программой einverted | Координаты предсказанных элементов вторичной структуры тРНК веб-сервисом RNAfold |
|---|---|---|---|
| Акцепторный стебель | 5'-С01-G07-3' (3'-C67-G73-5') | 5'-С01-G07-3' (3'-C67-G73-5') | 5'-С01-G07-3' (3'-C67-G73-5') |
| Дигидроуридиновый (D) стебель | 5'-C9-С13-3' (3'-G23-G27-5') | — | 5'-С9-С13-3' (3'-G23-G27-5') |
| Псевдоуридиновый (T) стебель | 5'-51C-54G-3' (3'-G62-65C-5') | — | 5'-51C-54G-3' (3'-G62-65C-5') |
| Антикодоновый стебель | 5'-A39-G43-3' (3'-C29-C33-5') | — | 5'-A39-G43-3' (3'-U29-C33-5') |
| Общее число канонических пар нуклеотидов | 21 | 7 | 21 |
Из приведённой таблицы нетрудно заметить, что программа einverted намного хуже справляется с поиском элементов вторичной структуры тРНК, так как принцип её работы заключается в поиске палиндромных последовательностей, в то время как принцип работы RNAfolds заключается анализе термодинамических характеристик предсказываемых структур.
Поиск ДНК-белковых контактов в заданной структуре.
В данном задании предлагалось провести анализ взаимодействий в выданной ДНК-белковой структуре (PDBI ID: 1OZJ).
Сначала необходимо было задать множества атомов кислорода 2'-дезоксирибозы, в остатке фосфорной кислоты, атомов азота в азотистых основаниях, а также пару скрипт-файлов, один из которых бы определял все перечисленные множества, а другой — последовательное изображение всей структуры, только ДНК в проволочной модели и той же модели, но с выделенными шариками множеством атомов кислорода и азота, определённые выше. Ниже приведены соответствующие команды в командной строке PyMOL:
select set1, name O3'+O4'+O5'
select set2, name OP1+OP2
select set3, name N1+N2+N3+N4+N6+N7+N9Ссылка на скрипт 1
Ссылка на скрипт 2
Далее проводилось сравнение контактов разных типов в ДНК-белковом комплексе. Будем считать полярными атомы кислорода и азота, а неполярными — атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5Å. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5Å.
| Остатки, с которыми наблюдается контакт атомов белка | Полярные контакты | Неполярные контакты | Всего контактов |
|---|---|---|---|
| 2'-дезоксирибоза | 0 | 9 | 9 |
| фосфорная кислота | 12 | 10 | 22 |
| Азотистые основания со стороны большой бороздки | 6 | 5 | 11 |
| Азотистые основания со стороны малой бороздки | 0 | 0 | 0 |
Судя по представленным в таблице данным, наиболее представлены конкакты между атомами белка и атомами остатка фосфата в нуклеотидах. Интересно, что совсем не представлены контакты атомов белка с атомами азотистых оснований со стороны малой бороздки, т.е. этот белок взаимодействует только с внешней бороздкой ДНК.
Затем была получена популярная схема ДНК-белковых контактов анализируемого комплекса (PDB ID: 1OZJ) с помощью программы nucplot. На вход данная программа принимает PDB-файл только в старом формате, поэтому скачанный файл структуры формат .pdb был предварительно конвертирован в подходящий формат с помощью программы remediator. Ниже приведены команды, используемые для получения схемы:
remediator --pdb --old 1OZJ.pdb > 1OZJ_old.pdb
nucplot 1OZJ_old.pdbПотом полученная схема была визуализирована с помощью ассоциированной программы GSview (см. Рис. 1., Рис. 2.).
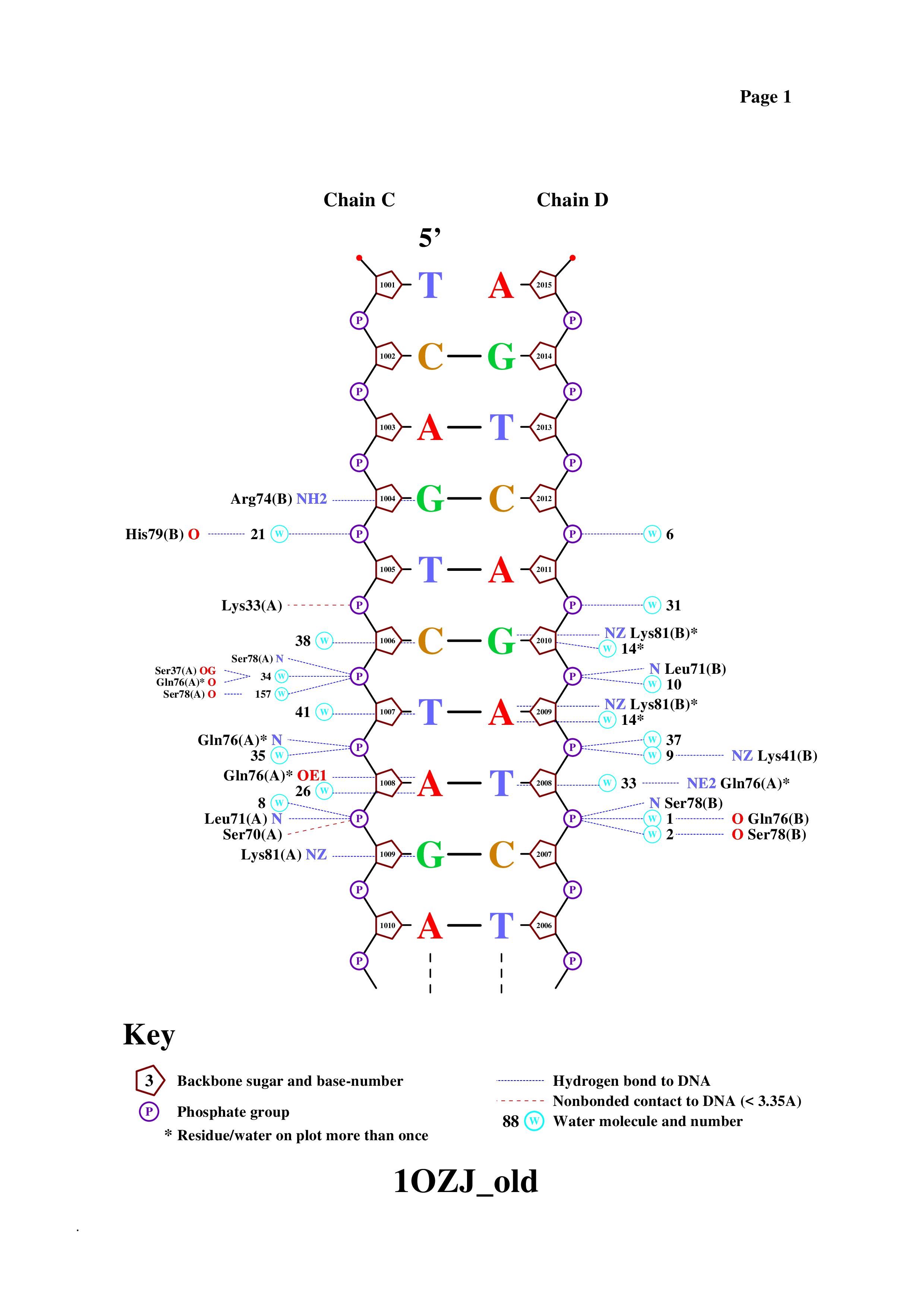
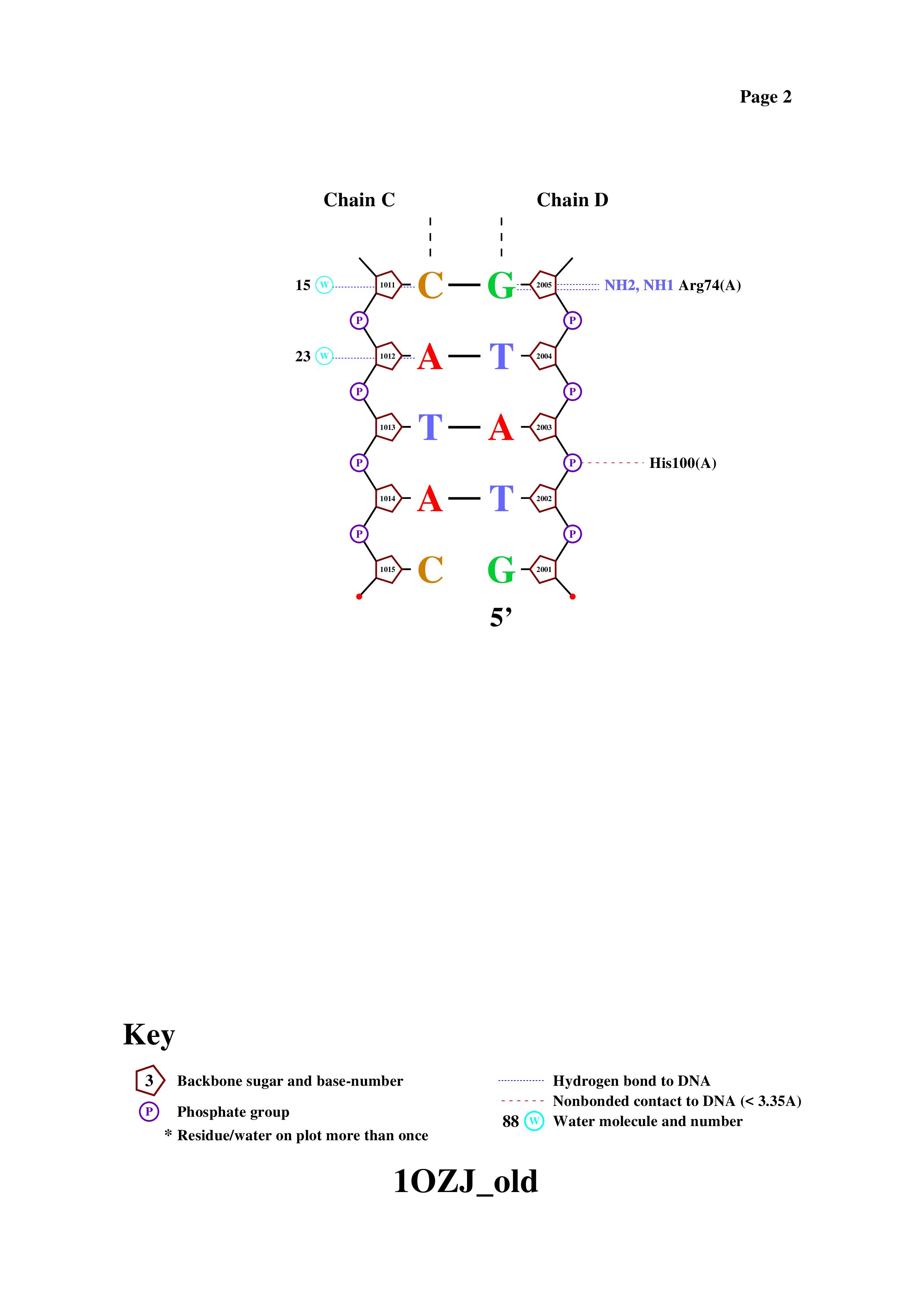
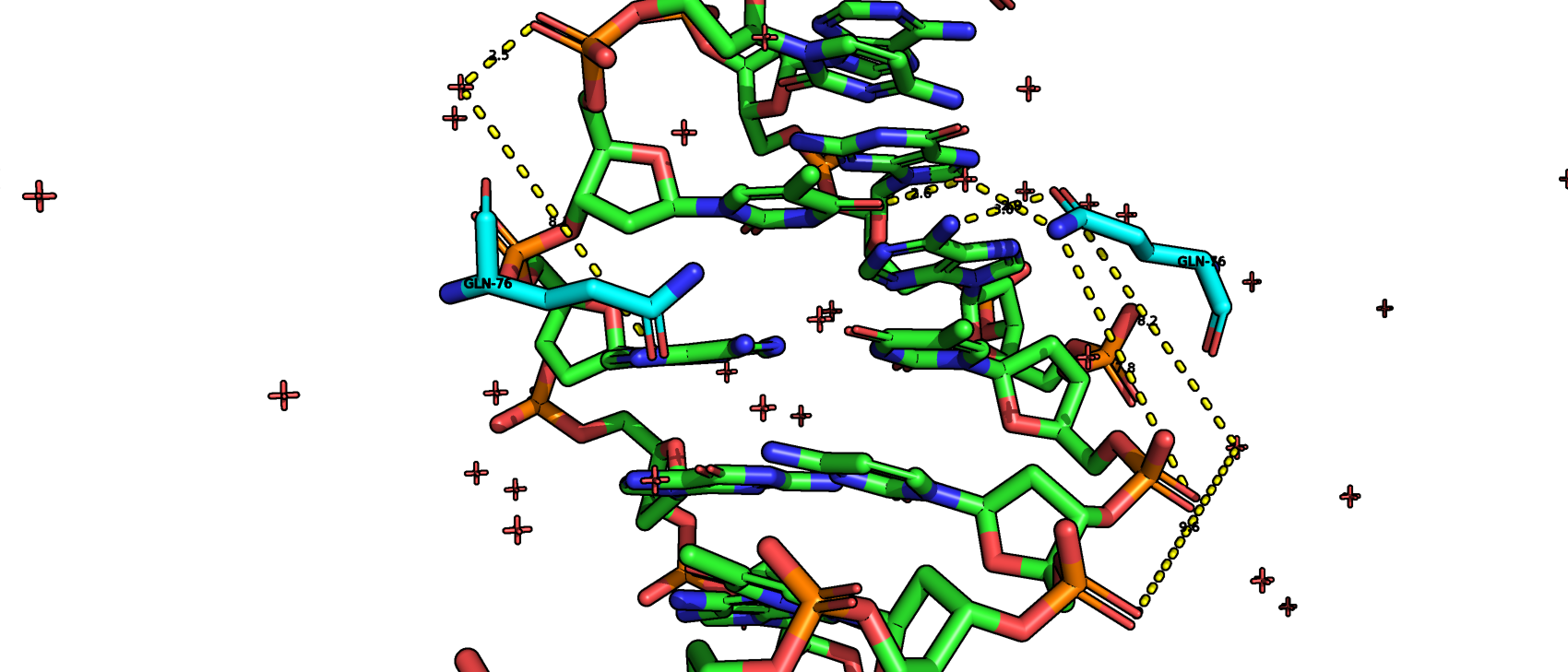
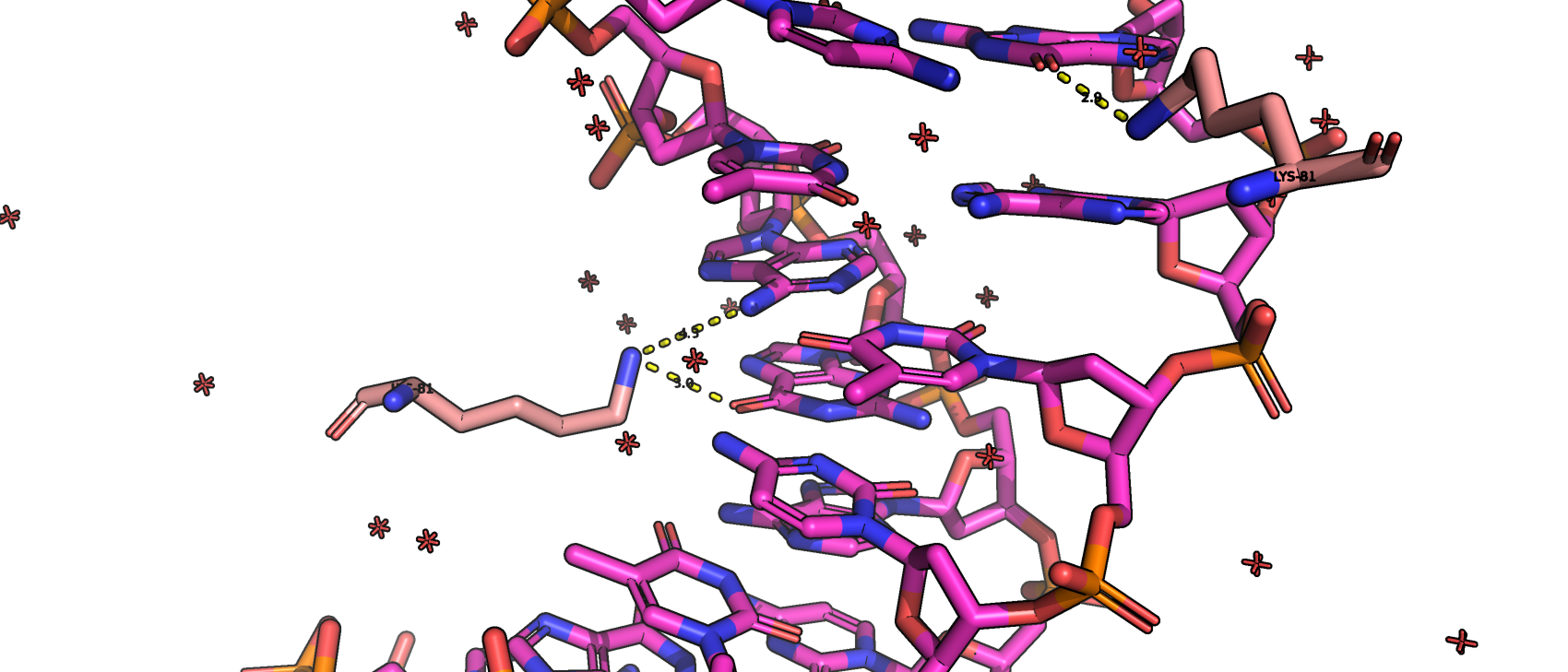
На представленной выше схеме наибольшим количеством связей (а именно пятью) с ДНК обладает 76 остаток глутамина (Gln76, см. Рис. 3.). Наиболее важным аминокислотным остатком для распознавания последовательности ДНК, на мой взгляд, является 81 лизин (Lys81), так как он образует целых 3 связи непосредственно с азотистыми основаниями (см. Рис. 4.).
The end...