HMM-профили и эволюционные домены
В заключительном практикуме этого семестра предлагалось построить HMM-профиль подсемейства какого-нибудь белкового семейства по последовательностям доменов и провести поиск этим профилем по всем белкам из выбранного семейства, чтобы рассчитать численные характеристики выделения подсемейства профилем и в целом оценить эффективность такого метода.
Для работы было выбрано семейство белков, кодирующих β-цепь натрий-калиевой АТФазы (AC: PF00287, name: Na_K-ATPase). Na+/K+-АТФаза — это фермент класса транслоказ, который осуществляет перенос катионов натрия во внешнюю среду, а калия — внутрь клетки, затрачивая на этот процесс энергию АТФ. Однако β-цепь является некаталитической субъединицей этого фермента и регулирует число натриевых насосов, доставляемых на плазматическую мембрану, путём регуляции сборки α/β-гетеродимеров фермента. Всего в этом семействе представлено 8436 белков. В seed содержится 55 последовательностей, в full — 6026.
Подсемейство было выделено по доменной архитектуре. Была выбрана такая архитектура, которая содержалась в показательном для проведения множественного выравнивания и дальнейшего построения профиля числе белков (см. Рис. 1.). Здесь репрезентативное число = 200-300 белков, т.е. было выбрано подсемейство, содержащее примерно такое количество белков — в моём случае 392 (если брать меньше, получится подсемейство с 10 белками, больше — только с 7935). Таким образом, выравнивание проводилось по 392 белковым последовательностям (см. Рис. 2.). Стоит отметить, что и выравнивание, и, соответственно, построение HMM-профиля проводились именно по домену, а не по всей белковой последовательности.


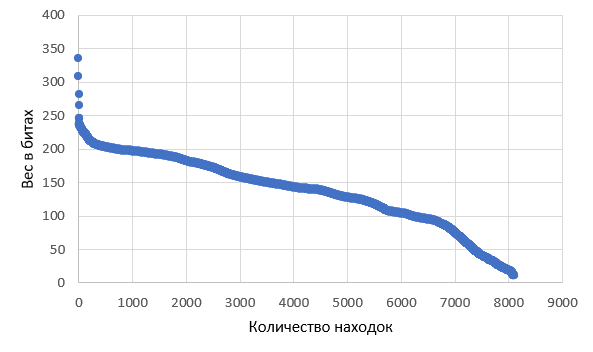
Всего из 8436 белков в базе было найдено 8100 статистически значимых хитов. Посмотрев на распределение весов (bit score) всех находок, можно увидеть весьма заметное увеличение угла наклона графика распределения в районе 7000-й находки (см. Рис. 3.). Следовательно, в этой области и будет находиться оптимальный порог на вес находки, который лучше всего выделяет подсемейство на фоне всего семейства. В данном случае было принято значение веса в 90 битов. Команды для построения профиля и поиска этим профилем по семейству белков приведены ниже.
hmmbuild --amino hmm_profile.hmm term4_pr10_MSA.fa
hmmsearch hmm_profile.hmm protein-matching-PF00287.fasta > hmmsearch_results.txt
В конце рассчитывались численные характеристики выделения подсемейства профилем (см. Таблица 1.). Всего находок, имеющих вес выше порогового, оказалось 6707. Истинно положительные значения ("True Positives", TP) в данном случае представляют собой все находки с весом более 90 битов и содержащие в своём названии "Sodium/potassium-transporting". Все остальные находки с весом более 90, но не содержащие такого имени (например, "Uncharacterized"), были признаны ложноположительными ("False Positives", FP). Их количество было вычислено с помощью следующей команды:
grep -v "Sodium/potassium-transporting" hmmsearch_results_table.txt | wc -lгде hmmsearch_results_table.txt — файл с табличной выдачей hmmsearch. Таких находок оказалось 1342, соответственно, TP = 6707 - 1342 = 5365.
Находки с весом меньше порогового, но входящие в рассматриваемое подсемейство белков, являются ложноотрицательными ("False Negatives", FN), их количество может быть вычислено как разница между суммарным количеством находок (8100) и суммой истинно и ложноположительных находок (8100 - (TP+FP) = 1393). Количество истинно отрицательных находок ("True Negatives", TN), то есть имеющих вес меньше порогового и не принадлежащих выделяемому подсемейству, вычисляется как разница между общим количеством белков в базе и суммарным количеством находок (8436 - 8100 = 336).
| TRUE | FALSE | |
|---|---|---|
| Positives | 5365 | 1342 |
| Negatives | 336 | 1393 |
Для оценки эффективности метода рассчитаем стандартные метрики Precision и Recall:
Precision = TP/(TP+FP) = 5365/(5365+1342) = 0.8 = 80%. Следовательно, профиль для подсемейства достаточно специфичен;
Recall = TP/(TP+FN) = 5365/(5365+1393) = 0.794 = 79.4%. Следовательно, профиль достаточно чувствителен для поиска белков подсемейства среди семейства.
Таким образом, описанный метод показал довольно высокую эффективность для выделения подсемейства белков на фоне соответствующего семейства.