Сигналы, мотивы и формы их представления
Описание мотива белка паттерном
Для выполнения задания были выбраны белки, содержащие в своих названиях мнемонику "SYR", которой обозначают аргинил-тРНК-синтетазу. Этот фермент осуществляет реакцию присоединения аминокислоты аргинина к соответствующей ему тРНК — аминоацилирование. Всего в банке белковых последовательностей бактерий из Swiss-Prot было найдено 670 белков с такой мнемоникой. Для поиска использовалась следующая команда на bash:
grep -c "SYR_" /P/y24/term4/bacteria-sw.fastaДалее проводилось выравнивание последовательностей 10 белков с такой мнемоникой в названии, выбранных произвольным образом. Названия белков были следующими: SYR_ECOLI, SYR_CORGL, SYR_STRPN, SYR_BACSU, SYR_LISW6, SYR_BURCH, SYR_MAGMM, SYR_YERE8, SYR_NEIMF, SYR_SHEAM. Ссылка на файл с выравниванием. Для создания паттерна был выбран участок выравнивания с 527 по 538 позицию (см. Рис. 1.). Затем по выбранному мотиву был составлен следующий паттерн:
Y-[MVAL]-[QL]-Y-[AG]-[YH]-[TA]-R-[IVL]-x-[SG]-[IVM].

Потом по полученному паттерну производился поиск в банке белковых последовательностей бактерий из Swiss-Prot с помощью команды fuzzpro:
fuzzpro -sequence /P/y24/term4/bacteria-sw.fasta -pattern "Y-[MVAL]-[QL]-Y-[AG]-[YH]-[TA]-R-[IVL]-x-[SG]-[IVM]" -outfile results_SYR.fuzzproПо результатам поиска было найдено 313 белков с выбранной мнемоникой, содержащих данный мотив, представленный в виде паттерна. Так как всего белков с такой мнемоникой в банке было найдено 670, то количество ложноотрицательных находок составило 357.
Нетрудно заметить, что метод поиска мотива по паттерну является малоэффективным, однако можно попытаться улучшить паттерн, например, заменив все выражения, содержащие 3 и более буквы в скобках, на x, а также убрав несколько малоконсервативных остатков с одного из концов паттерна. В итоге получился более оптимальный паттерн, по которому аналогично проводился поиск программой fuzzpro:
fuzzpro -sequence /P/y24/term4/bacteria-sw.fasta -pattern "Y-x-[QL]-Y-[AG]-[YH]-[TA]-R-x" -outfile results_SYR_fine-tuned.fuzzproЧисло находок в данном случае составило 476, что уже больше похоже на истинное значение.
Поиск мотивов в белках программой MEME и поиск этих мотивов в банке
Далее происходил поиск мотивов программой MEME со следующими опциями: последовательности аминокислотные, по одному представителю мотива на последовательность, минимальная длина мотива 8, максимальная длина мотива 15 остатков, не более трёх мотивов на выравнивание:
meme SYR_sequences.fasta -protein -minw 8 -maxw 15 -mod oops -nmotifs 3Здесь SYR_sequences.fasta — файл, содержащий последовательности каждого из выбранных ранее 10 белков (не выравнивание!). На выходе была получена директория meme_out, содержащая файл meme.html. По результатам работы программы было найдено 3 мотива во всех выбранных белках с мнемоникой (см. Рис. 2.).
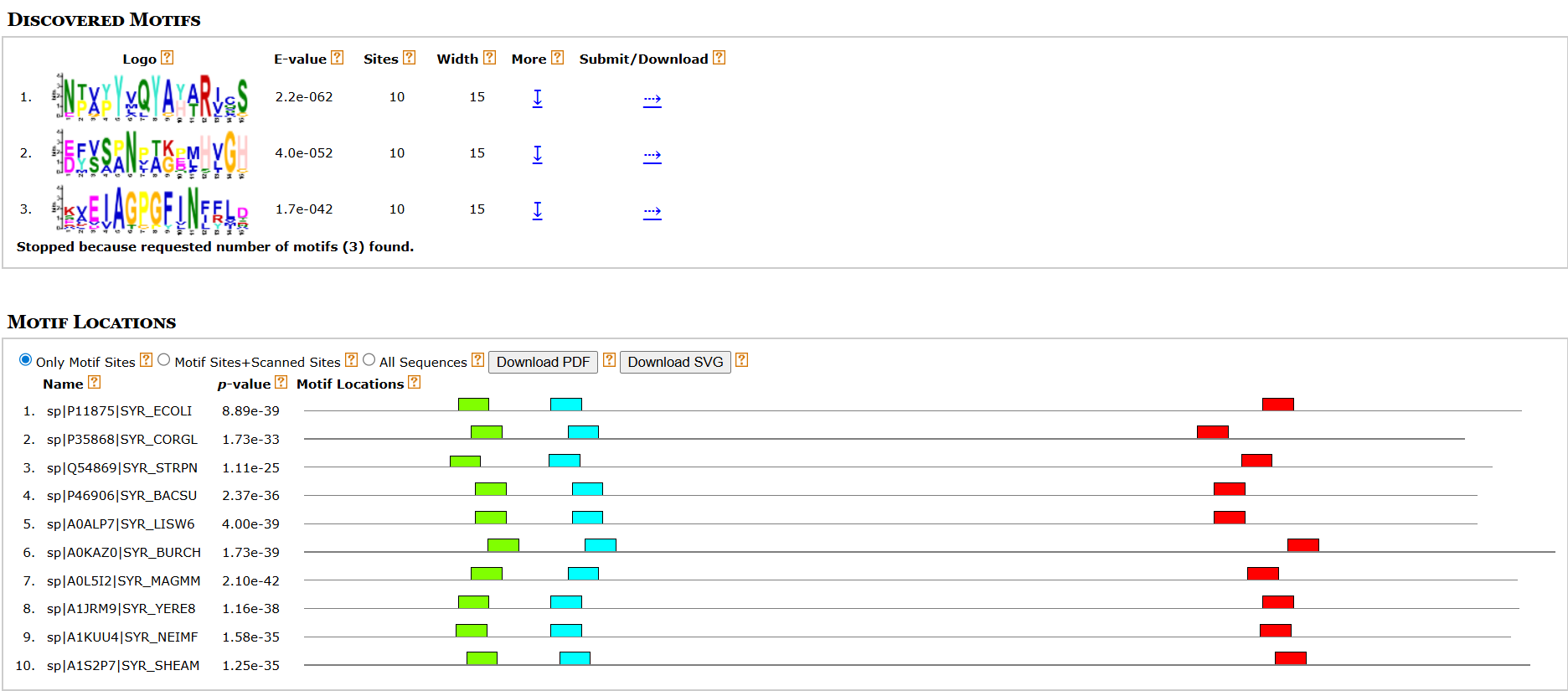
mast public_html/term4/pr8/meme_out/meme.html /P/y24/term4/bacteria-sw.fastaНа выходе был получен файл mast.html.
В результате с помощью программы MAST найденные программой MEME мотивы были обнаружены во всех 670 белках, содержащих соответствующую мнемонику (см. Рис. 3.).
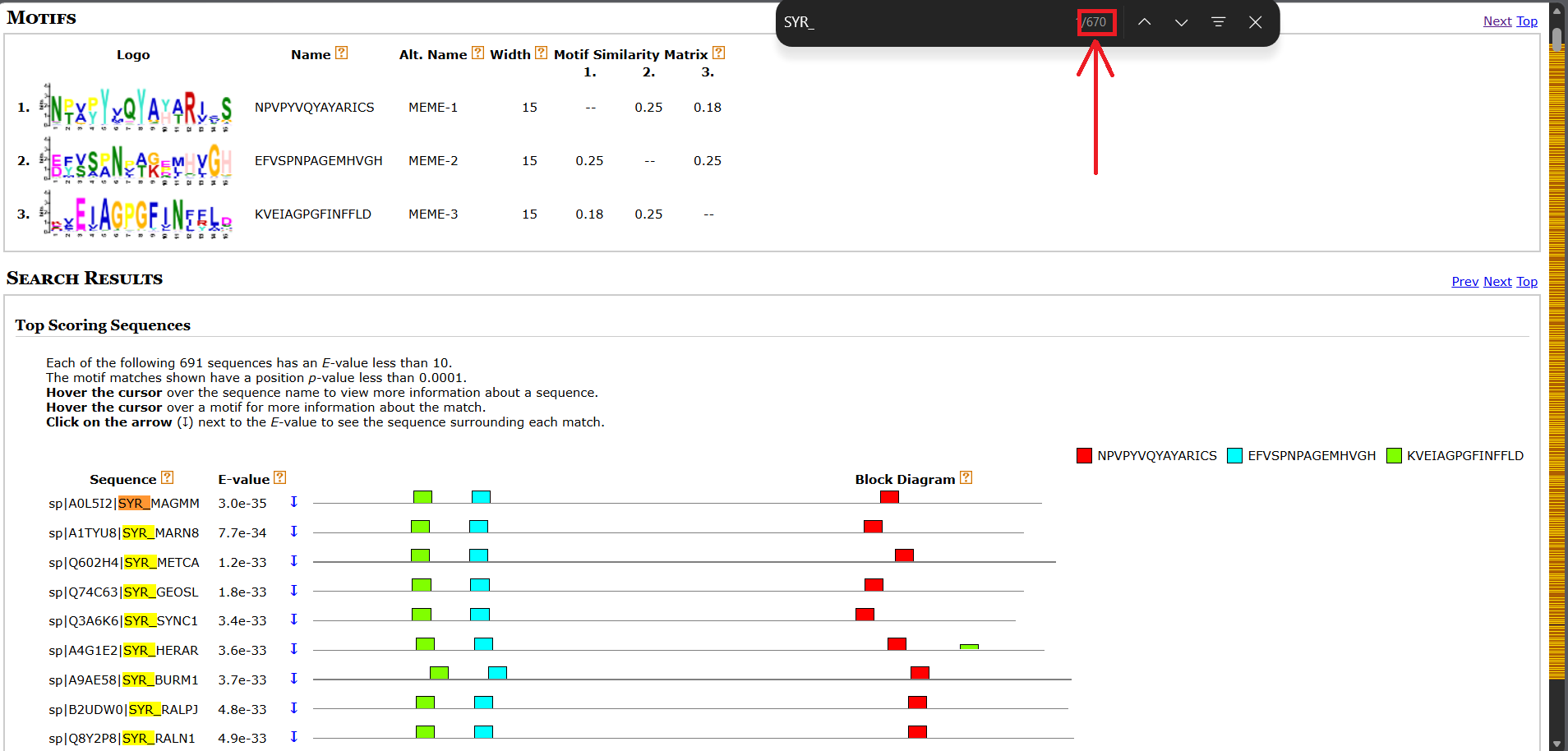
Стоит также добавить, что такая высокая точность поиска мотивов в банке последовательностей обусловлена тем, что программа MEME представляет мотивы в виде позиционных весовых матриц (PWM), что позволяет учитывать вариативность аминокислот в каждой позиции мотива белка.
Поиск последовательности Шайна-Дальгарно в геноме прокариота
В конце предлагалось провести поиск последовательности Шайна-Дальгарно (ПШД) в полной последовательности генома прокариота, выбранного в первом семестре для написания мини-отчёта. В моём случае выбранным прокариотом является архея Natronomonas pharaonis, геном которой представлен одной кольцевой хромосомой и двумя кольцевыми плазмидами. С помощью программы fuzznuc производился поиск ПШД, представляющей собой мотив AGGAGG, по обеим (прямой и комплементарной) цепям геномной ДНК:
fuzznuc -sequence public_html/term1/genome/GCF_000026045.1_ASM2604v1_genomic.fna -pattern AGGAGG -outfile straight_strand_SD.fuzznuc
fuzznuc -sequence public_html/term1/genome/GCF_000026045.1_ASM2604v1_genomic.fna -pattern AGGAGG -complement Y -outfile all_strands_SD.fuzznuc
grep -v "+" all_strands_SD.fuzznuc > reverse_strand_SD.fuzznucВсего было обнаружено 825 находок на прямой цепи (790 на хромосоме и 25 на плазмидах) и 1611 на обратной цепи (1540 на хромосоме и 71 на плазмидах).
Учитывая то, что длина генома археи составляет 2749696 пар оснований, а вероятность встретить ПШД (шестинуклеотидный мотив) составляет приблизительно (1/4)^6, то по случайным причинам
ожидается получить 2749696*(1/4)^6 ≈ 670 находок. Для прямой цепи значение числа находок подозрительно близко к числу случайных совпадений, что, по всей видимости,
говорит об отсутствии статистической значимости этого результата. Для обратной цепи ситуация значительно лучше, однако, учитывая тот факт, что рассматриваемая архея содержит
около 2800 генов в своём геноме, такие значения без привязки к конкретным генам (вероятность встретить подобный короткий мотив в произвольном месте гена или в промежутке между
генами довольно велика) также с трудом можно считать достоверными.
Просмотрев глазами координаты 20 случайно выбранных находок и сравних их с координатами предполагаемых ПШД, исходя из таблицы геномных особенностей
(ПШД должна находиться на расстоянии 5-15 нуклеотидов от старт-кодона CDS), не было обнаружено ни одной последовательности, которую бы действительно можно было
считать истинной ПШД. Очевидно, это говорит о чрезвычайно низкой эффективности такого подхода для поиска функциональных
последовательностей, так как, задав строгий консенсус мотива без его предварительного соотнесения с участками CDS, на которых теоретически должны располагаться
ПШД, мы упускаем из виду как возможную вариативность мотива (которая у прокариот весьма значительна), так и привязку к определённым функционально значимым участкам генов,
что не даёт значимого результата для данной задачи.