Занятие 4
1. Построение дерева по нуклеотидным последовательностям
В задании требовалось построить филогенетическое дерево тех же бактерий, что в предыдущих заданиях, используя последовательности РНК малой субъединицы рибосомы (16S rRNA).
В таблице для каждой бактерии представлены AC записи EMBL, в которой нашлась последовательность 16S rRNA, координаты этой РНК в этой записи, на прямой или комплементарной последовательности оказалась последовательность этой рРНК.
| Название | Мнемоника | AC записи EMBL | Координаты РНК | Цепь |
| Bacillus anthracis | BACAN | AE015927 | 8715..10233 | Обратная |
| Clostridium tetani | CLOTE | AE016879 | 9335..10841 | Прямая |
| Finegoldia magna | FINM2 | AP008971 | 611796..613319 | Прямая |
| Geobacillus kaustophilus | GEOKA | BA000043 | 10421..11973 | Прямая |
| Lactobacillus acidophilus | LACAC | CP000033 | 59255..60826 | Прямая |
| Lactococcus lactis | LACLM | AM406671 | 511423..512971 | Прямая |
| Staphylococcus aureus | STAA1 | AP009324 | 531922..533476 | Прямая |
| Streptococcus pyogenes | STRP1 | AE004092 | 17170..18504 | Прямая |
Последовательности были выровнены программой Muscle (with Defaults) в JalView. Затем выравнивание было импортировано в программу MEGA, с помощью которой было реконструировано филогенетическое дерево методом Neighbor Joining. Полученное и правильное деревья представлены на рисунках:
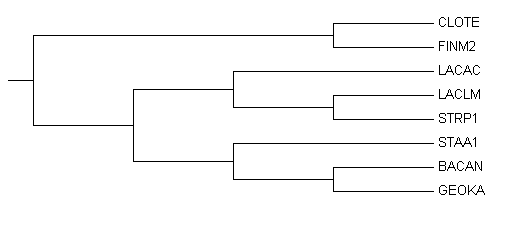
|
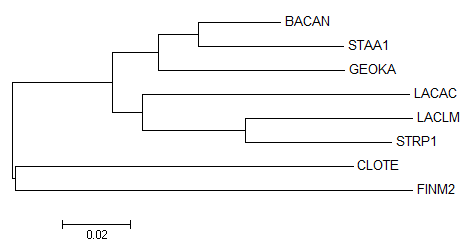
|
| Верное дерево | Neigbour joining tree of 16S rRNA |
Реконструированное дерево очень похоже на правильное. Единственное отличие - в нем есть ветвь {BACAN, STAA1} vs {GEOKA, STRP1, LACLM, LACAC, FINM2, CLOTE} вместо {BACAN, GEOKA} vs {STAA1, STRP1, LACLM, LACAC, FINM2, CLOTE}. Теоретически качество дерева, построенного по нуклеотидным последовательностям, должно быть хуже, чем построенного по белкам, потому что одну аминокислоту можно закодировать разными кодонами, и в белковых выравниваниях не будут учитываться молчащие мутации в нуклеотидах. Думаю, белок, который я выбрала в предыдущем практикуме - фактор элонгации трансляции G (EFG) - был менее удачным.
2. Построение и анализ дерева, содержащего паралоги
В задании требовалось найти в своих бактериях достоверные гомологи белка CLPX_BACSU, построить дерево этих гомологов. Считая дерево реконструированным верно, указать несколько пар ортологов и несколько пар паралогов. Привести примеры отражённых на дереве эволюционных событий двух типов: 1) дупликация гена; 2) разделение путей эволюции белков в результате видообразования.
Два гомологичных белка будем называть ортологами, если они а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования.
Два гомологичных белка из одного организма будем называть паралогами.
Чтобы найти гомологов в заданных организмах, воспользовались файлом proteo.fasta, где лежат записи банка UNIPROT, относящиеся к бактериям, перечисленным в таблице к заданию 1. Провели поиск гомологов программой blastp(с порогом на E-value 0,001) и отобрали по мнемонике видов только те находки, которые относятся к отобранным ранее бактериям (файл с выравниваниями blastp.out).
Использовались следующие команды:makeblastdb -in proteo.fasta -dbtype prot -out prot blastp -query clpx_bacsu.fasta -db prot -out blastp.out -evalue 0.001 -outfmt 6
Чтобы отобрать белки нужных организмов можно воспользоваться утилитой awk:
for id in bacsu clote bacan geoka lacac laclm staa1 strp1;
do awk '{ print $2 }' blastp.out | grep -i $id >> ids.txt; done
Список белков в файле ids.txt. Список последовательностей приведен в файле homologs_seqs.fasta
С помощью программы muscle выровняли последовательности. Команда:
muscle -in homologs_seqs.fasta -out alignment.fasta
Построили дерево в программе MEGA методом Neighbor Joining:
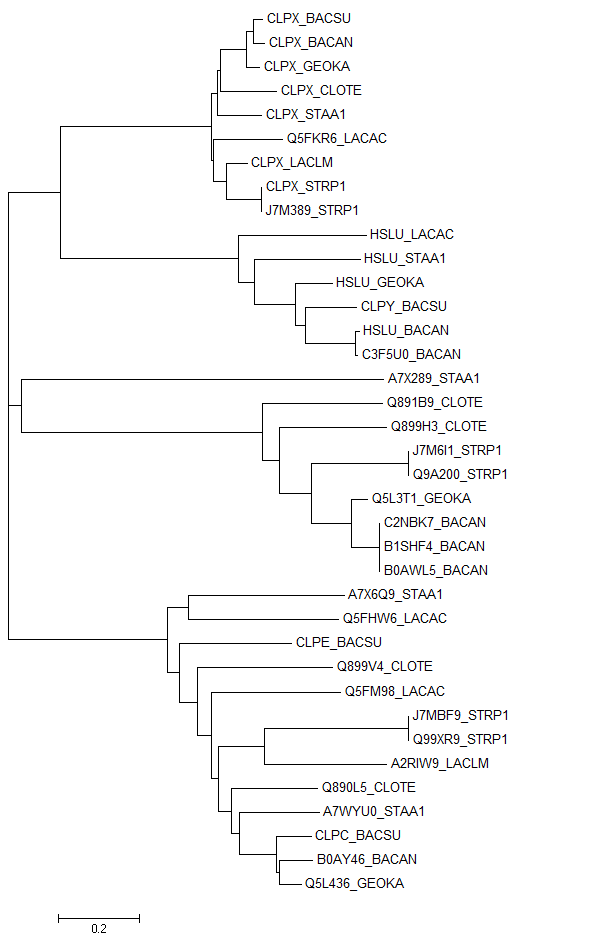
Среди найденных гомологичных белков есть ортологи:
CLPX_BACSU и CLPX_BACAN; HSLU_LACAC ,HSLU_STAA1, HSLU_GEOKA, HSLU_BACAN
И паралоги:
Q891B9_CLOTE и Q899H3_CLOTE, Q5FM98_LACAC и Q5FHW6_LACAC
Примеры:
- дупликация гена
- разделение путей эволюции белков в результате видообразования
J7MBF9_STRP1 и Q99XR9
HSLU_GEOKA и CLPY_BACSU