Задание d1. Определение вторичной структуры
Определение вторичной структуры
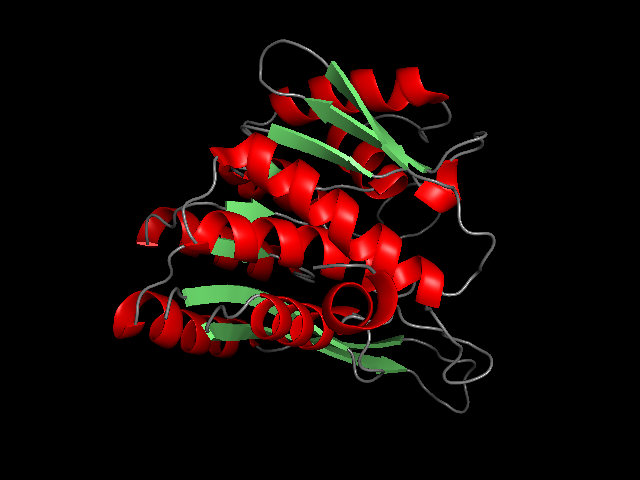
Для определения вторичной структуры 2I5B использовалась программа DSSP. Выдача программы: -2I5B.dssp-. На рисунке ниже изображены элементы вторичной структуры цепи А 2I5B.
В таблице 1 приведено сравнение результата выдачи программы для четырех α-спиралей и четырех β-тяжей
| № элемента вторичной структуры | Результат DSSP (номера остатков) | Аннотация в pdb-файле (номера остатков) | |
| α-спираль | |||
| 1 | 19 – 27 | 18 – 30 | |
| 2 | 59 - 66 (67 - 73 516 спираль) | 58 – 73 | |
| 3 | 86 - 98 | 85 – 99 | |
| 4 | 121 - 126 (127 - 131 516 спираль) | 120 – 131 | |
| β-тяж | |||
| 1 | 51 – 56 | 52 – 56 | |
| 2 | 33 – 45 | 33 – 44 | |
| 3 | 5 - 13 | 5 – 13 | |
| 310 спирали | |||
| 1 | 132 - 134 | 132 – 135 | |
| 2 | 180 - 182 | 180 – 183 | |
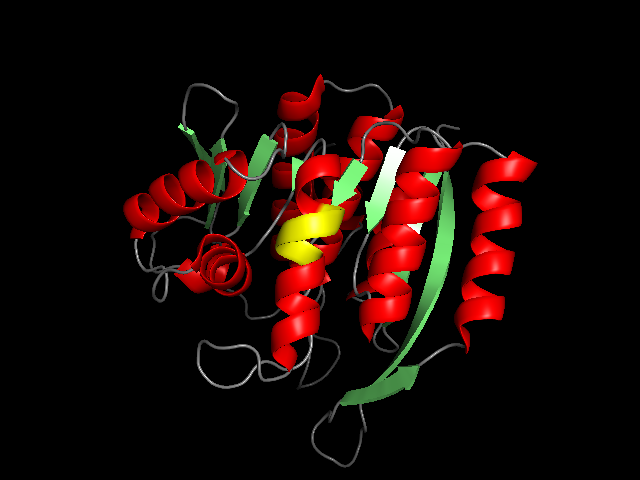
В целом, границы, определенные с помощью DSSP и в аннотации PDB-файла схожи. Можно увидеть, что границы β-листов совпадают или отличаются на один остаток, границы же α-спиралей меньше соответствуют PDB-файлу. Любопытно,что для двух спиралей (остатки 58 – 73 и 120 – 131 по аннотации PDB) программой DSSP было определено, что их части, 67 - 73 и 127 - 131 соответственно, являются 516, а не α-спиралями. В аннотации PDB это не отмечено. Спираль 120 – 131 показана ниже, участок 127 – 131 покрашен желтым цветом.
Построение карты β-листа с помощью программы SheeP
С помощью программы SheeP построили карту β-листа цепи А структуры 2I5B. Изображение одной из карт показано ниже.
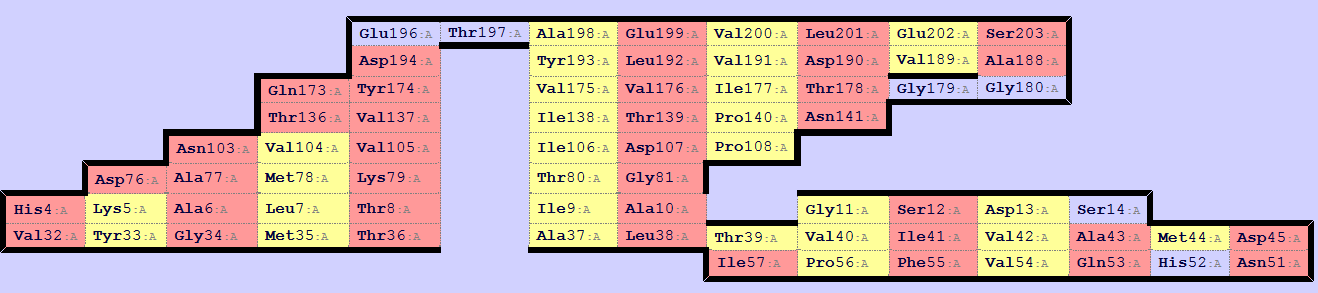
На рисунке ниже изображен β-лист, показанный на карте. Он состоит из 9 тяжей.
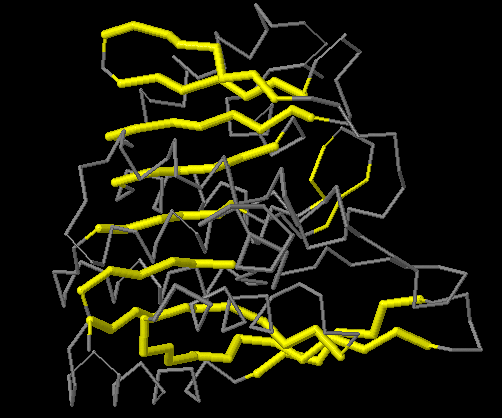
Столбец в карте соответствует "хребту" β-листа. На рисунке ниже показан хребет, соответствующий столбы с аминокислотами 198, 193, 175, 138, 106, 80, 9, 37.
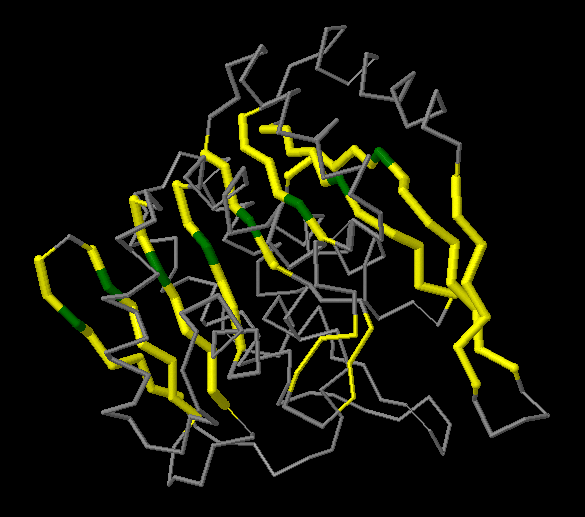
К гидрофобному ядру обращена "желтая" сторона листа. Это будет более наглядно видно в задании d3.
Программа Stride умеет предсказывать элементы вторичной структуры, а также выдает список водородных связей. Файл с выдачей программы 2i5b_hbonds.txt. Для карты водородных связей выбрали следующий участок, состоящий из трех тяжей и четырех гребней:
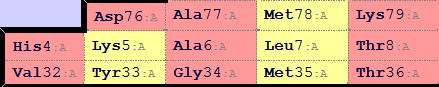
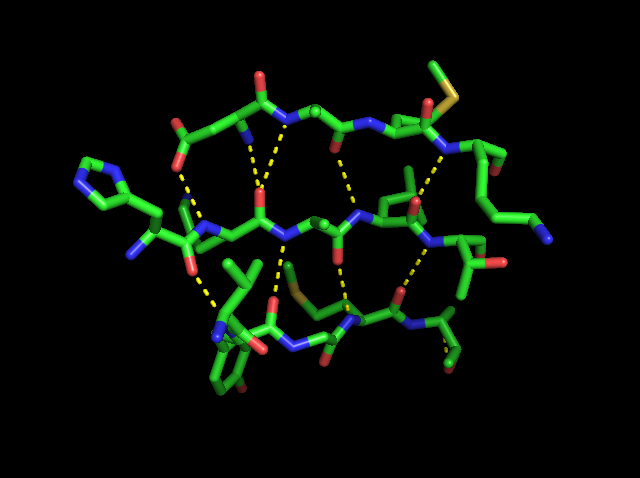
По выдаче программы Stride построили карту водородных связей участка (ориентация участка на всех рисунках сохранена):
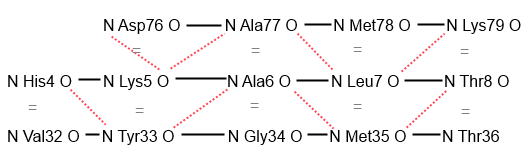
Есть один участок нерегулярности: Lys5 выступает как акцептор и образует две водородные связи - с Asp76 и Ala77.
Задание d2. Совмещение структур
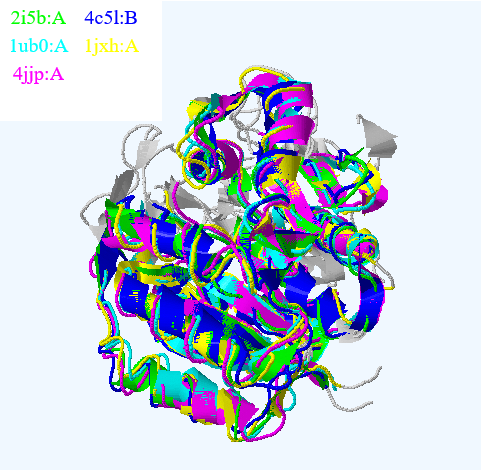
Построение совмещения белка pyridoxine kinase (2I5B) и четырех гомологов
С помощью сервиса PDBeFold было найдено 4 гомолога 2I5B: 4C5L, 1UB0, 1JXH и 4JJP (Искали для цепи А). На рисунке ниже показано совмещение структур.
Структурное выравнивание superpos.fasta (большие буквы в файле соответствуют выровненным позициям) можно сравнить с выравниванием по последовательностям seq_alignment.fasta (выполнено в JalView TCoffee со стандартными параметрами).

На рисунке ниже показан участок, на котором выравнивания по последовательностям и по структурам не совпадают. Можно увидеть, что по структурам все аспарагиновые кислоты 192 позиции выравниваются, но по последовательностям аспарагиновые кислоты двух структур- нет (190 позиция выравнивания).

Если обратиться к наложению пространственных структур, то можно увидеть, что аспартаты действительно довольно хорошо выравниваются, поэтому скорее структурное выравнивание правильное.
Поиск по сходству структур в PDBeFold
В PDBeFold осуществили поиск структурных гомологов домена 1dek A: 33-154. Использовали параметры по умолчанию:
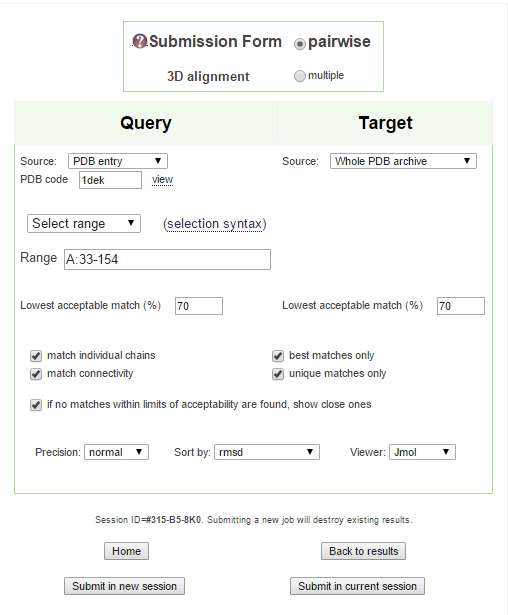
Выдача была отсортирована по RMSD. В ней 35 структур (PDBeFold_output.txt), исходного домена в ней нет. Дело в том, что параметры по умолчанию требуют того, чтобы процент совпадения структур был не меньше 70, а так как домен, который мы ищем, занимает меньше 70% общей длины белка, белок не находится.
Совмещение структур альфа- и бета- цепочек константного домена T-клеточного рецептора
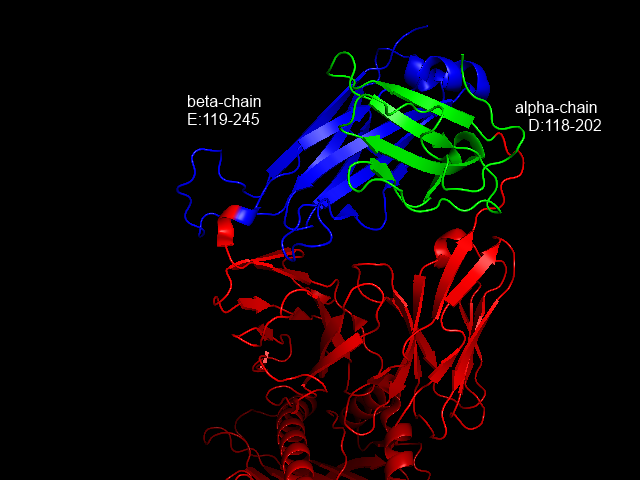
Была выбрана структура Т-клеточного рецептора 1OGA: альфа-цепь - region d:118-202, бета-цепь - region e:119-245. Альфа- и бета-цепи показаны на рисунке:
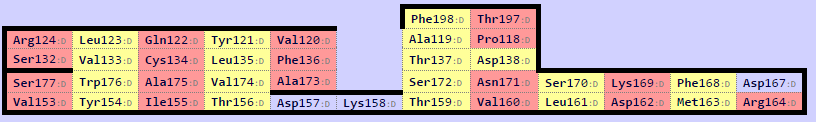
С помощью SheeP построили карту бета-листов:
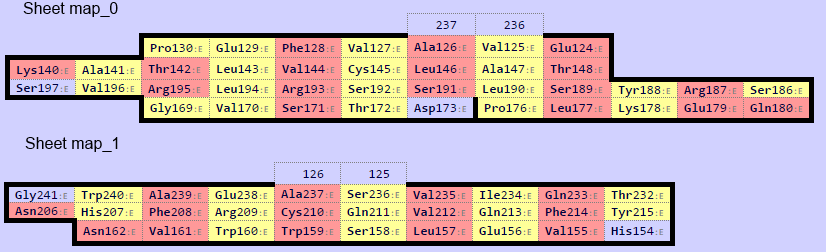
Бета-листу альфа-цепочки соответствует первый бета-лист бета-цепочки. На рисунках выше они изображены в одной ориентации. Консервативный остаток цистеина - 145 в бета-цепочке и 134 в альфа-цепочке.
Сомещение цепочек было построено в Pymol с помощью следующих команд:
pair_fit \ 1oga_alpha & resi 134 & n. ca, 1oga_beta & resi 145 & n. ca, \ 1oga_alpha & resi 122 & n. ca, 1oga_beta & resi 127 & n. ca, \ 1oga_alpha & resi 175 & n. ca, 1oga_beta & resi 192 & n. ca, \ 1oga_alpha & resi 155 & n. ca, 1oga_beta & resi 172 & n. ca, \ 1oga_alpha & resi 133 & n. ca, 1oga_beta & resi 144 & n. ca, \ 1oga_alpha & resi 135 & n. ca, 1oga_beta & resi 146 & n. ca
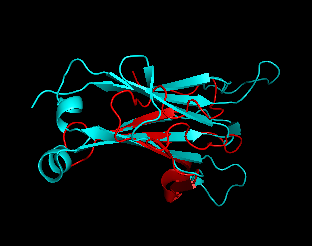
RMSD совмещения - 0,485. Общий ход полипептидной цепи совпадает. Совмещение сохранено в файле.
Задание d3. Нахождение гидрофобных кластеров
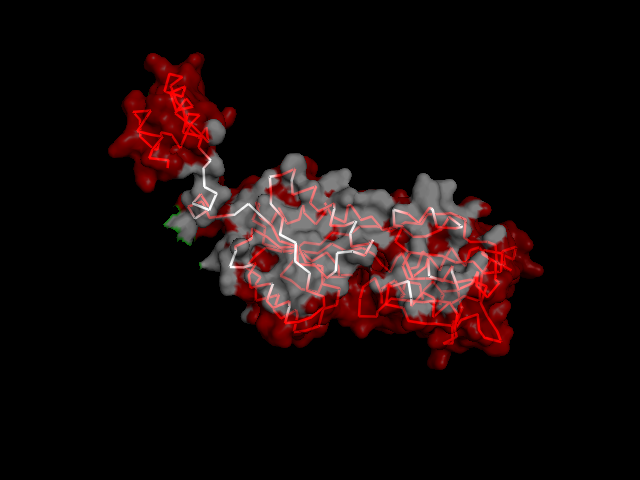
С помощью программы Clud были найдены гидрофобные кластеры в структуре 2I5B. С порогом расстояния 5 и минимальным размером кластера 7 было получено 4 гидрофобных ядра, которые показаны на рисунках ниже. В белке есть одно очень крупное ядро, состоящее из 2504 атомов, принадлежащих одной стороне β-листа и части α-спирали, к которая обращена к этой стороне. Другие гидрофобные ядра принадлежат некоторым петлям и части β-листа.

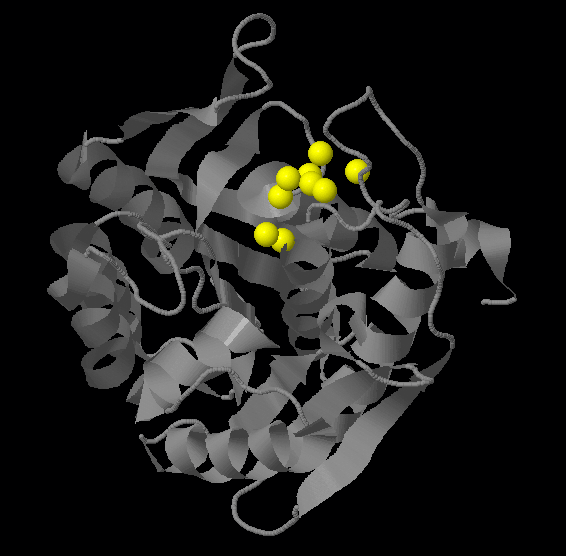
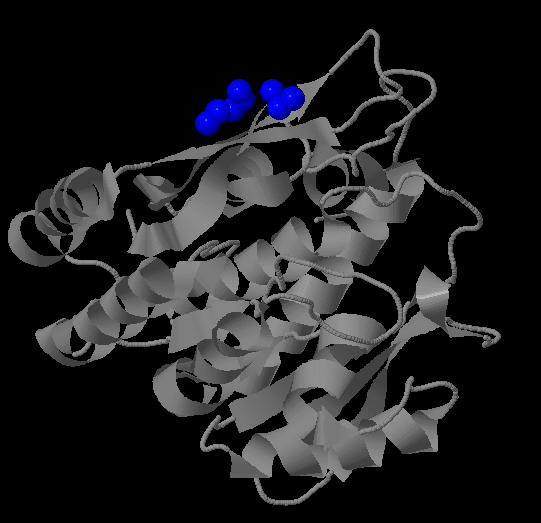
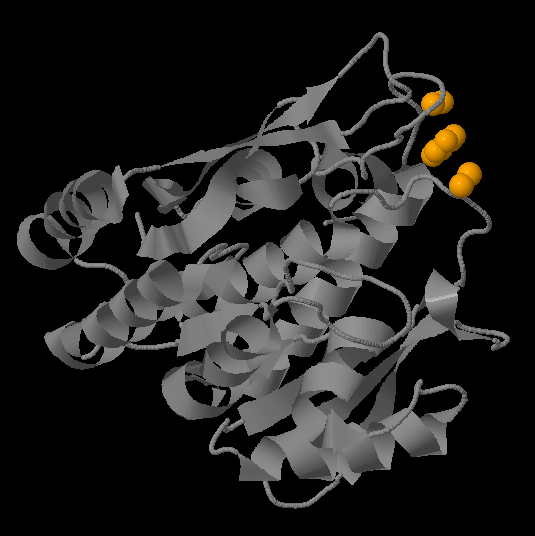
Самый большой гидрофобный кластер соответствует единственному фосфометилпиримидинкиназному домену белка (позиции 13-261).
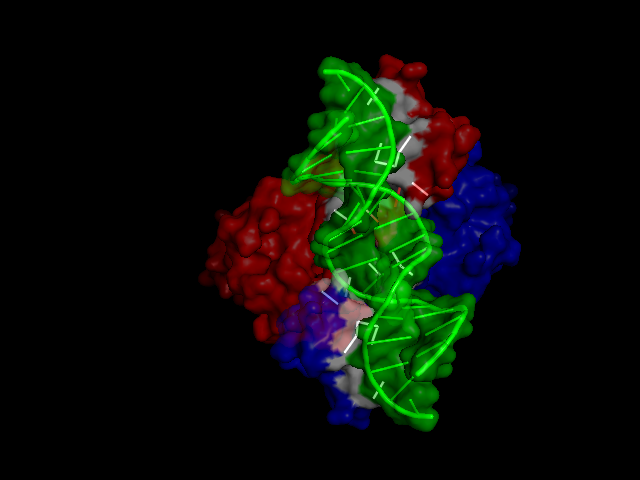
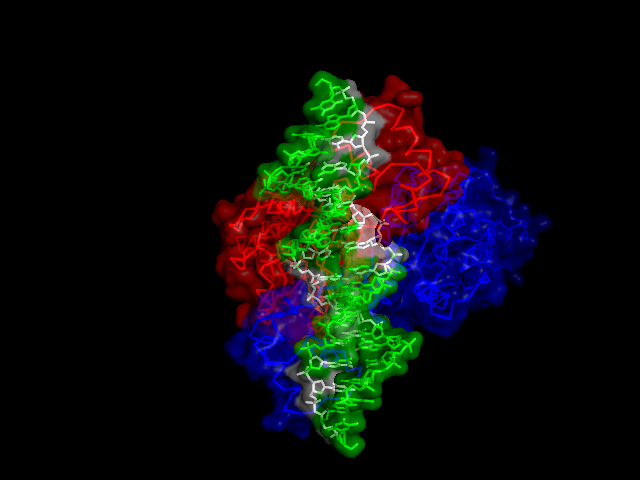
Также нашли гидрофобные ядра комплекса белка с ДНК, принадлежащего семейству ETS. С порогом расстояния 5.4 и минимальным размером кластера 5 было найдено 3 кластера: большой кластер, состоящий из 95 атомов, принадлежащих α-спиралям, кластер, состоящий из 20 атомов, принадлежащим частям α-спиралей и петли, и небольшой кластер из 6 атомов, принадлежащий части спирали, взаимодействующей с ДНК. Изображения кластеров представлены ниже.
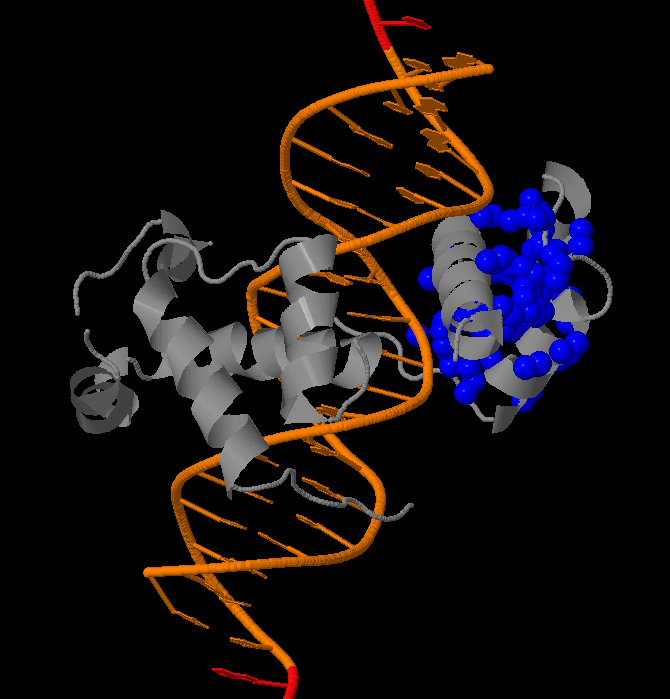
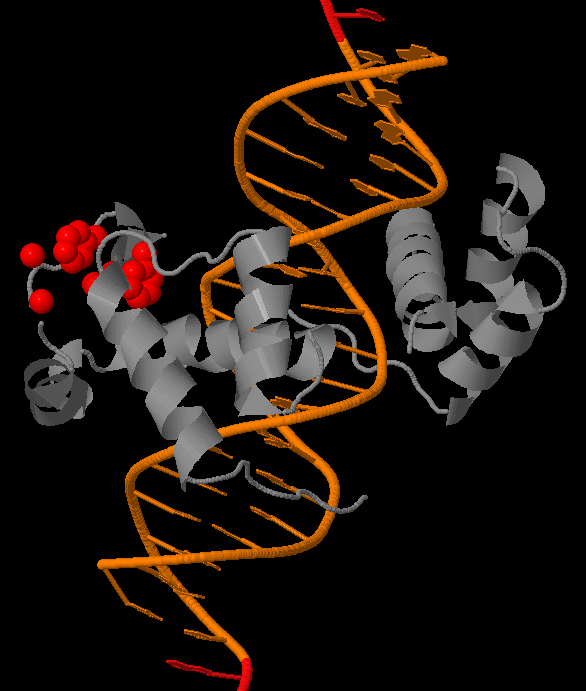
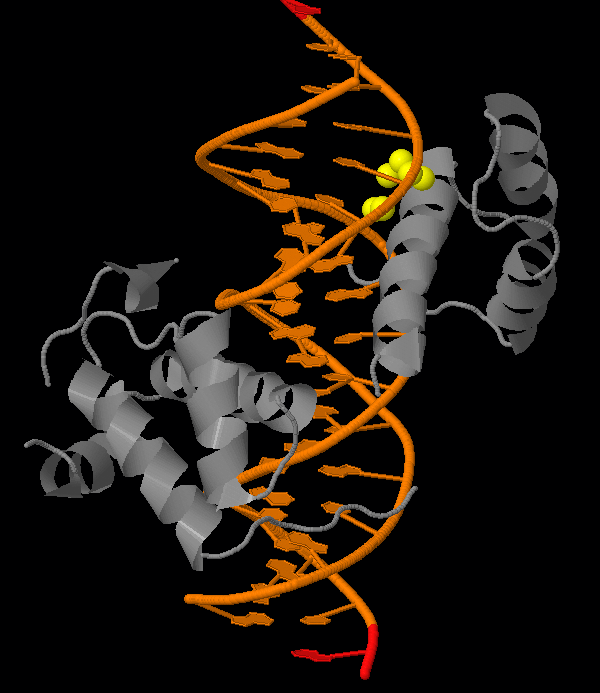
Задание d4: Построение поверхности, раскраска участка поверхности: pymol
Получение изображений поверхностей контактов c помощью pymol
Для выполнения задания была выбрана структура 1qp7 ДНК-связывающего белка, репрессора транскрипции PurR типа спираль-поворот-спираль. Изображения поверхности контакта мономера белка с симметричным мономером, контакта димера белков с двойной спиралью и контакта ДНК с димером белков, полученные средствами pymol, показаны на рисунках ниже.
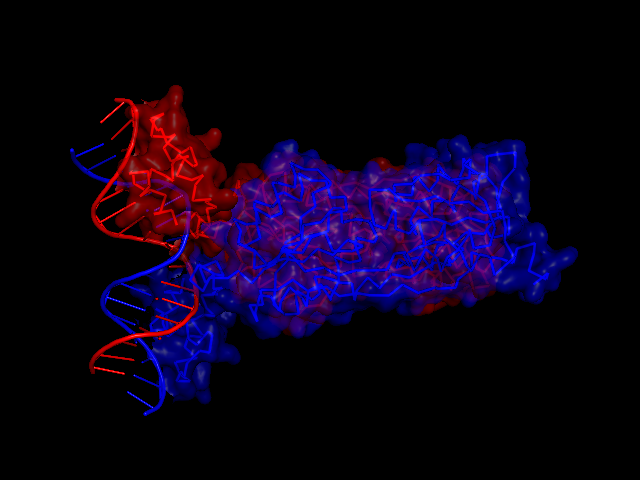
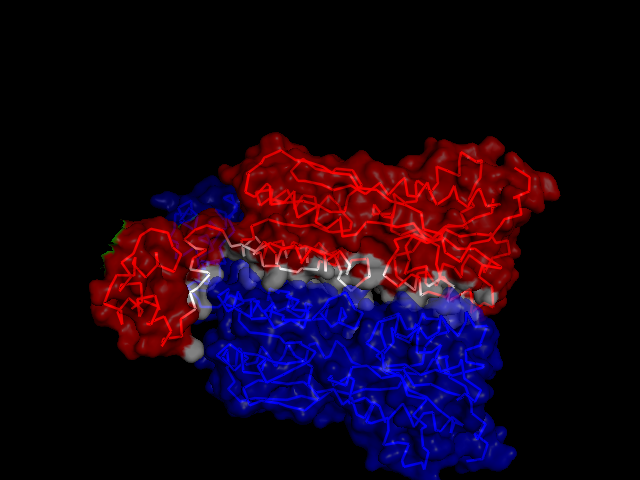
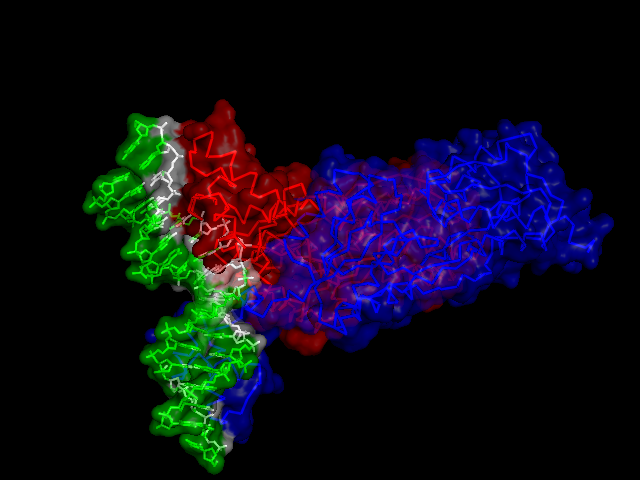
Изображение гидрофобных поверхностей (CluD)
С помощью следующих команд сохранили в файл координаты димера 1qp7:
load 1QP7.pdb, A_1 create B_1, A_1 alter_state 1,B_1,(x,y,z)=(x,-y,-z) save 1QP7_dimer.pdb
С помощью сервиса Clud было найдено 4 гидрофобных кластера размером не менее 10 атомов:
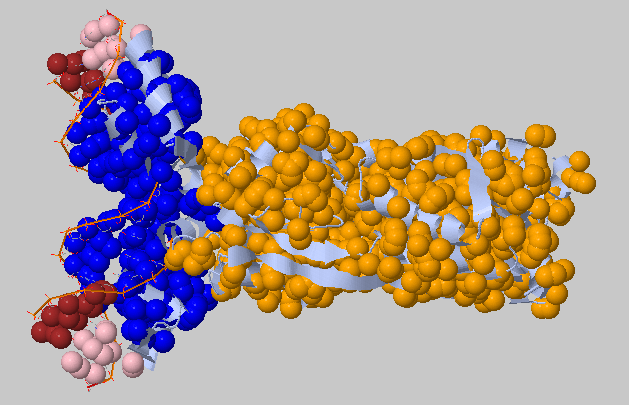
Изображения поверхностей контактов мономеров белков, образующих контакты с другим мономером, а также поверхности контакта ДНК с димером белков показаны на рисунке ниже. Синим цветом выделена поверхность, относящаяся к атомам, входящим в найденные гидрофобные кластеры.
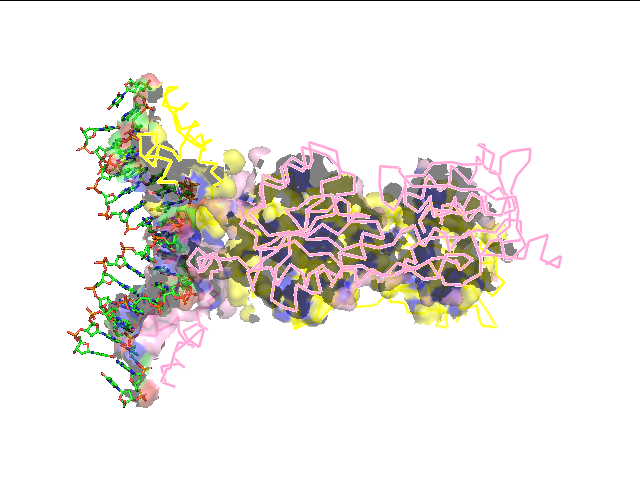
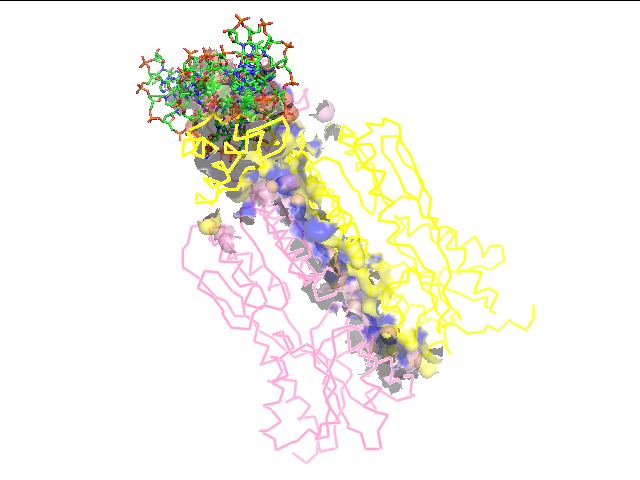
Задание d5: Сравнение доменов SCOP/SCOPe, ECOD, CATH и Pfam
Для транскрипционного репрессора PurR HTH-типа(UniProt ID - PURR_ECOLI, PDB ID - 1QP7) сравнили границы доменов согласно базам данных SCOP/SCOPe, ECOD, CATH и Pfam.
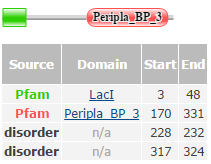
Согласно Pfam в белке два домена: LacI (3-48) и Peripla_BP_3 (170-331).
По SCOP также два домена, N-терминальный домен d1qp7a2: 1qp7 с границами 3-58:
| Класс | All alpha proteins |
| Фолд | lambda repressor-like DNA-binding domains |
| Суперсемейство | lambda repressor-like DNA-binding domains |
| Семейство | GalR/LacI-like bacterial regulator |
И С-терминальный d1qp7a2: 1qp7 с границами 59-340:
| Класс | Alpha and beta proteins (a/b) |
| Фолд | Periplasmic binding protein-like I |
| Суперсемейство | Periplasmic binding protein-like I |
| Семейство | L-arabinose binding protein-like |
По CATH в белке три домена:
- 1qp7A01 с границами 3-59 из суперсемейства lambda repressor-like DNA-binding domains
- 1qp7A02 с границами 60-161 и 292-323 и топологией укладка Россмана
- 1qp7A03 с границами 162-291 и 324-340 и топологией укладка Россмана
Согласно ECOD (Evolutionary Classification of Protein Domains) в белке три домена:
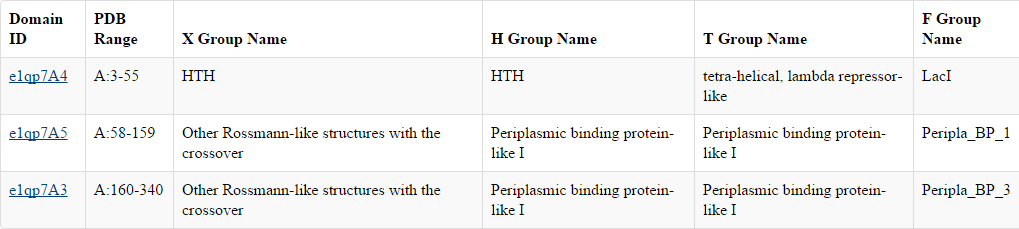
Таким образом, границы доменов и даже их число для одной структуры в разных базах данных довольно различается.
Задание d6: Использование сайта PDB
Получение последовательностей белков, структуры которых определены с помощью электронной микроскопии
Для того, чтобы получить последовательности белков, стуктуры которых определены с помощью метода электронного микроскопии, воспользовались advanced search на сайте PDB, а именно : Experimental Method, ELECTRON MICROSCOPY. Было обнаружено 917 структур. Результат поиска в fasta-формате: el_micr.fasta
Поиск пары белков, гибкое структурное выравнивание включает большее число атомов, чем жесткое
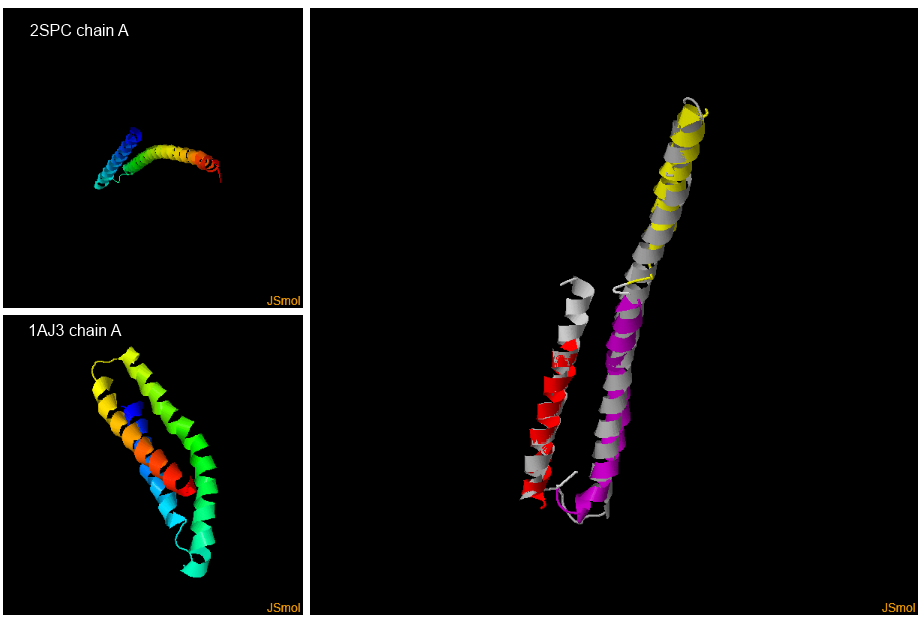
В качестве пары были выбраны структуры спектрина 2spc, цепь A (107 остатков) и альфа-спектрина 1aj3, цепь А (98 остатков) (Взяты из статьи по FATCAT 2003 года).
- Число позиций выравнивания: 67
- RMSD: 4.6
- p-value: 1.38e-06
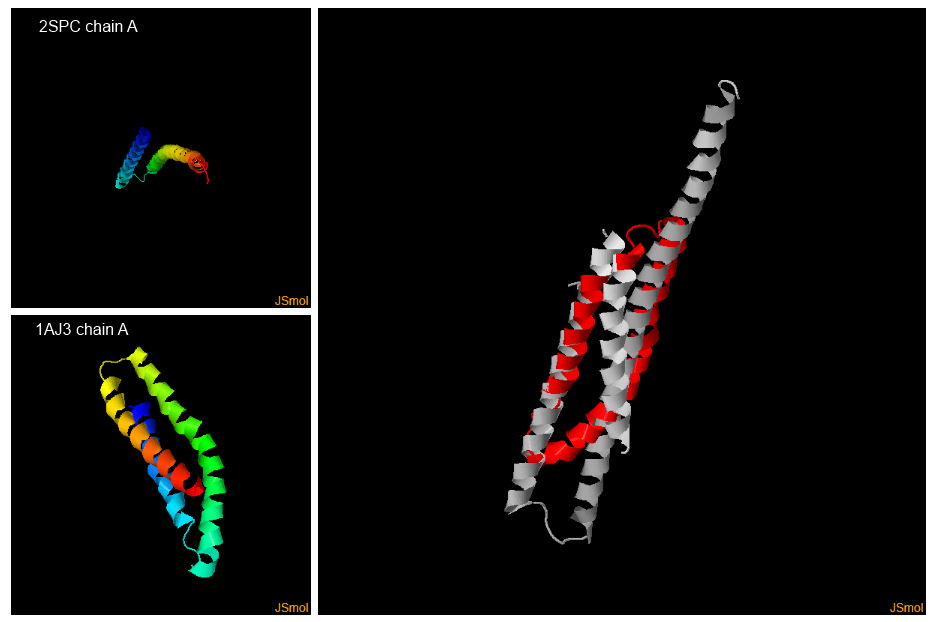
Гибкое выравнивание было выполнено с помощью программы FATCAT:
- Число позиций выравнивания: 94
- RMSD: 2.2 (сделано 2 твиста)
- p-value: 1.05e-06