На главную страницу второго семестра
Глобальное и локальное выравнивание аминокислотных последовательностей
Матрицы переходов
-
Глобальное выравнивание
-Последовательности:
1) MIKK - первые 4 остатка BTUB_ECOLI
2) MAWKT - 5 аминокислот полученных следующим образом:
в последовательности первых 4-х остатков BTUB_ECOLI проведены две замены и в произвольном месте вставлена аминокислота
-Параметры построения матрицы перехода
Вес совпадения=2
Вес замены=-1
Штраф за делецию=-2
-Матрица переходов
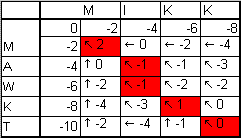
-Выравнивание оптимального пути
-Вес оптимального пути = 0.
-
Локальное выравнивание
-Последовательности:
1) MIKKASLLT - первые 9 аминокислот BTUB_ECOLI
2) IKLLT - аминокислоты 2, 3, 7, 8, 9 из BTUB_ECOLI
-Параметры построения матрицы перехода
Вес совпадения=2
Вес замены=-1
Штраф за делецию=-2
-Матрица переходов
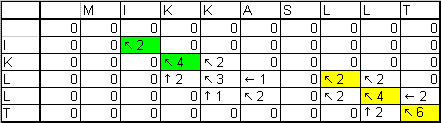
-Пути выравнивание:
оптимальный

|
субоптимальный

|
-Вес пути
| оптимального = 6 |
субоптимального = 4 |
Влияние параметров на глобальное выравнивание
Ниже приведены два выравнивания белка BTUB_Ecoli c последовательностью, сотканной из двух участков, 11-ти- и 12-тибуквенных, того же белка.Разницей в параметрах я вляется только штраф за открытие делеции. При построении выравнивания применялась команда needle из пакета EMBOSS.
Выравнивание 1.
- Параметры:
-
Штраф за открытие делеции - 1.0
- Штраф за продление делеции - 1.0
-
Результат:
- Идентичность - 2.9%
- Cхожесть - 3.1%
- Вес - 84
- Делеций - 22
|
1 MIKKASLLTACSVTAFSAWAQDTSPDTLVVTANRFEQPRSTVLAPTTVVT 50
1 0
51 RQDIDRWQSTSVNDVLRRLPGVDITQNGGSGQLSSIFIRGTNASHVLVLI 100
1 0
101 DGVRLNLAGVSGSADLSQFPIALVQRVEYIRGPRSAVYGSDAIGGVVNII 150
1 0
151 TTRDEPGTEISAGWGSNSYQNYDVSTQQQLGDKTRVTLLGDYAHTHGYDV 200
1 0
201 VAYGNTGTQAQTDNDGFLSKTLYGALEHNFTDAWSGFVRGYGYDNRTNYD 250
1 0
251 AYYSPGSPLLDTRKLYSQSWDAGLRYNGELIKSQLITSYSHSKDYNYDPH 300
1 0
301 YGRYDSSATLDEMKQYTVQWANNVIVGHGSIGAGVDWQKQTTTPGTGYVE 350
||||||||||| . | |. : |
1 MKQYTVQWANN--T-H-------------TV--S-Y-- 17
351 DGY-DQRNTGIYLTGLQQVGDFTFEGAARSDDNSQFGRHGTWQTSAGWEF 399
| | |.|
18 D-YVDAR 23
400 IEGYRFIASYGTSYKAPNLGQLYGFYGNPNLDPEKSKQWEGAFEGLTAGV 449
24 23
450 NWRISGYRNDVSDLIDYDDHTLKYYNEGKARIKGVEATANFDTGPLTHTV 499
24 23
500 SYDYVDARNAITDTPLLRRAKQQVKYQLDWQLYDFDWGITYQYLGTRYDK 549
24 23
550 DYSSYPYQTVKMGGVSLWDLAVAYPVTSHLTVRGKIANLFDKDYETVYGY 599
24 23
600 QTAGREYTLSGSYTF 614
24 23 |
Выравнивание 2.
- Параметры:
-
Штраф за открытие делеции - 10.0
- Штраф за продление делеции - 1.0
-
Результат:
- Идентичность - 2.1%
- Cхожесть - 2.4%
- Вес - 59
- Делеций - 1
|
1 MIKKASLLTACSVTAFSAWAQDTSPDTLVVTANRFEQPRSTVLAPTTVVT 50
1 0
51 RQDIDRWQSTSVNDVLRRLPGVDITQNGGSGQLSSIFIRGTNASHVLVLI 100
1 0
101 DGVRLNLAGVSGSADLSQFPIALVQRVEYIRGPRSAVYGSDAIGGVVNII 150
1 0
151 TTRDEPGTEISAGWGSNSYQNYDVSTQQQLGDKTRVTLLGDYAHTHGYDV 200
1 0
201 VAYGNTGTQAQTDNDGFLSKTLYGALEHNFTDAWSGFVRGYGYDNRTNYD 250
1 0
251 AYYSPGSPLLDTRKLYSQSWDAGLRYNGELIKSQLITSYSHSKDYNYDPH 300
1 0
301 YGRYDSSATLDEMKQYTVQWANNV-IVGHGSIGAGVDWQKQTTTPGTGYV 349
|||||||||||. .|.:..:.|.
1 MKQYTVQWANNTHTVSYDYVDAR 23
350 EDGYDQRNTGIYLTGLQQVGDFTFEGAARSDDNSQFGRHGTWQTSAGWEF 399
24 23
400 IEGYRFIASYGTSYKAPNLGQLYGFYGNPNLDPEKSKQWEGAFEGLTAGV 449
24 23
450 NWRISGYRNDVSDLIDYDDHTLKYYNEGKARIKGVEATANFDTGPLTHTV 499
24 23
500 SYDYVDARNAITDTPLLRRAKQQVKYQLDWQLYDFDWGITYQYLGTRYDK 549
24 23
550 DYSSYPYQTVKMGGVSLWDLAVAYPVTSHLTVRGKIANLFDKDYETVYGY 599
24 23
600 QTAGREYTLSGSYTF 614
24 23
|
Первое, что бросается в глаза - это резкая разница в гепах. В результате резкого увеличения штрафа, мы получили более "короткое" выравнивание, т.е. программа попыталась максимально сблизить аминокислоты мутантной последовательности, но на самом деле, в идеале, программа должна поставить вторую часть на то место, с которого она была снята, так же как поступила и с первой, однако для этого необходимо поставить ещё меньшее значение штрафа за открытие делеции. Вот почему в первом выравнивании (с таким безумным количеством делеций) схожесть и идентичность имеют более высокие значения.
©
Борщёв Иван, 2005