 Ралдугина Василиса Студентка Факультета биоинженерии и биоинформатики МГУ имени М.В. Ломоносова  |
Обо мне |
| |||||||||||||||||||||||||||||||
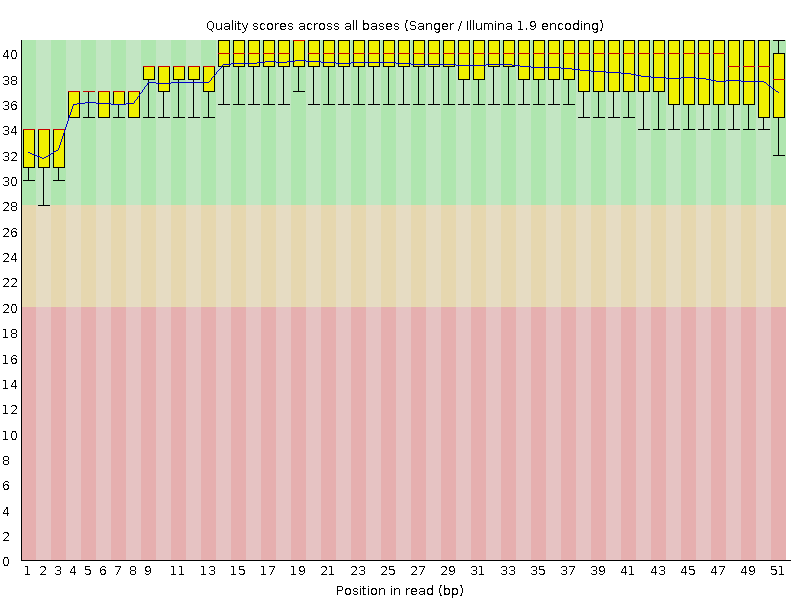
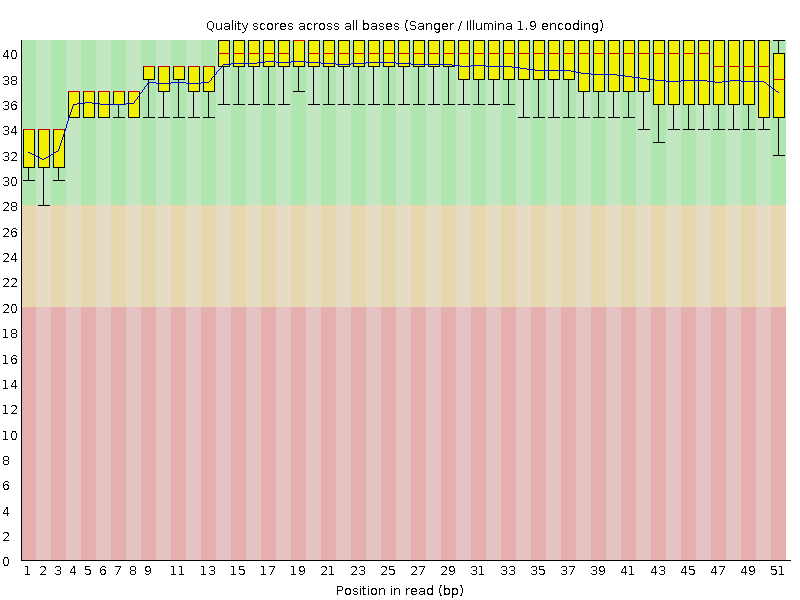
| Команда | Функция | Выдача |
| hisat2-build chr9.fasta chr9 | Индексирование референсной последовательности | Индексированный файл chr9.fasta |
| hisat2 -x chr9 -U out_chr9.1.fastq --no-softclip > chr9.sam | Выравнивание чтений после чистки с референсной последовательностью | Файл, который содержит выравнивание формата SAM 1_align.sam |
Мы не использовали параметр запрещающий без разрывов картировать, так как в данном случае у нас данные транскриптомного анализа, то есть мы работаем с РНК. По сравнению с ДНК с ней могли произойти модификации связанные с перегруппировкой (сплайсинг).
Далее необходимо было проанализировать, полученное выравнивание. Для этого я использовала программу Samtools. Она работает с файлами в формате SAM.
| Команда | Функция | Выдача |
| samtools view chr9.sam -bo chr9.bam | Программа переводит файл в формат bam | chr9.bam |
| samtools sort chr9.bam -T nexact.txt -o chr9_sort.bam | Сортировка выравнивания чтений и референса по координате в референсе | chr9_sort.bam |
| samtools index chr9_sort.bam | Индексирование отсортированного выравнивания | chr9_sort.bam |
| samtools idxstats chr13_sort.bam > result.txt | Запись числа откартировавшихся чтений | result.txt |
Выяснилось, что на хромосому откартировалось 18924 рида. 343 рида не были откартированы.
Часть III. Подсчет чтений.
Для подсчета чтений мы использовали пакет Bedtools.
Далее была использована функция bamtobed, которая позволяет из bam файла сделать bed:
Команда запуска: /P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtobed -i chr9.bam > chr9.bed
Выдача: chr9.bed
Затем нашли пересечение наших выравниваний с разметкой по генам:
Команда запуска: /P/y14/term3/block4/SNP/bedtools2/bin/bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b chr9.bed -c > chr.intersect.bed
Выдача: chr.intersect.bed
После этого для упрощения чтения файла была проведена сортировака:
Команда запуска: sort -k 6 -r us.bed > s.bed
Выдача: s.bed
После проведения подсчета чтений мы видим, что прочтения легли в границы двух генов. Прочтения, которые не попали скорее всего относятся к не матричной РНК.
Мои чтения относятся к GL877870 и CICP10
Ген CICP10 - псевдоген репрессора capicua 10. Длина этого гена 466 пар оснований. Координаты гена: 243,062,007-243,062,293
Ген GL877870. Длина этого гена 66021 пара оснований. Координаты гена: 243,059,660-243,189,373
Часть IV. Функции пакета BEDTOOLS
Задание 1. Получите из файла c выравниванием файл с чтениями в формате fastq
Команда для запуска:/P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtofastq -i chr9.bam -fq chr2.1.fastq
Выдача: chr2.1.fastq
Задание 4. Объедините Ваши чтения в кластеры.
Команда для запуска: /P/y14/term3/block4/SNP/bedtools2/bin/bedtools cluster -i chr9.bed -s >cluster.bed
Выдача: cluster.bed
Задание 3. Разбейте свою хромосому на фрагменты по 1 млн нуклеотидов. Какова длина хромосомы в нуклеотидах? Сколько в результате получилось интервалов?
Исходные файлы: chr9.txt. (содержание: chr9 141213431 )
Команда для запуска:/P/y14/term3/block4/SNP/bedtools2/bin/bedtools makewindows -g chr9.txt -w 1000000 > win.bed
Выдача: win.bed
Флаг -g - файл с указанием размера участков генома, w- размер промежутка. Длина хромосомы 141 213 431 п.н.. Получилось 142 промежутка.
![]()
![]()
![]()
© Raldugina Vasilisa 2016