Практикум 9.
Шаг 1. Выбор домена
Выбор пал на домен LAP1_C (PF05609). LAP1 (Lamina-associated polypeptide 1), содержащие этот домен, - белки с единственным трансмембранным участком. Локализуются на внутренней части ядерной мембраны. Они взаимодействуют и активируют торсин A, AAA+ АТФазу, локализованную в эндоплазматическом ретикулуме через перинуклеарный домен, и образуют гетерогексамерное кольцо (LAP1-Torsin)3, которое направляет торсин во внутреннюю часть ядерной мембраны. Схема анализируемой архитектуры представлен ниже.

Шаг 2. Анализ
Необработанный список белков с был скопирован из описания архитектуры. Список последовательностей всех белков скачан с пункта "Alignments". Далее список был приведен в более удобный вид и отфильтрован по данной архитектуре при помощи следующего скрипта.
#!/usr/bin/env python3
from Bio import SeqIO
import pandas
allseq = SeqIO.parse('full.fasta', 'fasta')
with open ('domain_organisation.txt', 'r') as names,
open('domain_organisation_list.txt', 'w') as names_only,
open('domain_organisation.fasta', 'w') as outf:
a = names.readlines()
for i in range(len(a)):
k=a[i].split()
a[i]=k[0]
names_list = a
names_only.write('\n'.join(names_list))
for i in allseq:
if i.name in names_list:
SeqIO.write(i, outf, "fasta")
Последовательности из полученного fasta-файла были выровнены следующей командой
muscle -in domain_organisation.fasta -out align.fasta
Далее полученное выравнивание обрезала до границы архитектуры в Jalview. Там же удалила странно выровнявшиеся и очень похожие последовательности (Edit => Remove redundancy, порог - 95). Далее на основании полученного файла искала архитектуру во всех последовательностях.
hmm2build HMM align_cut.fa hmm2calibrate HMM hmm2search -E 0.1 --cpu 1 HMM full.fasta > hmmresult.txt
Выходной файл анализировала следующим скриптом.
#!/usr/bin/env python3
from Bio import SeqIO
import pandas
import matplotlib.pyplot as plt
allseq = SeqIO.parse('pr9/full.fasta', 'fasta')
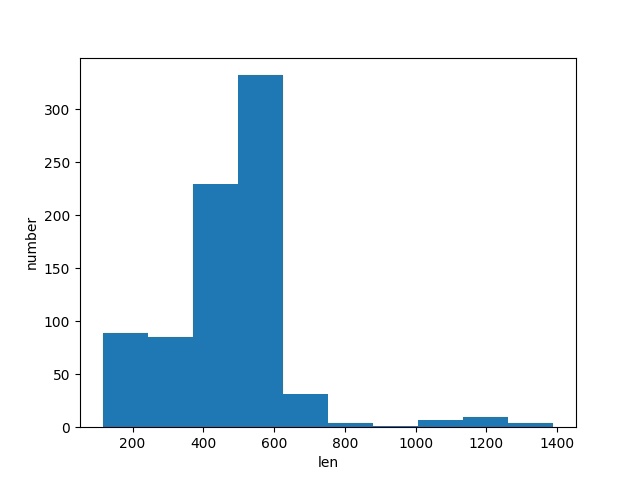
len_lst = [len(i.seq) for i in allseq]
fig = plt.figure()
ax = fig.add_subplot(1, 1, 1)
ax.hist(len_lst)
ax.set_ylabel('number')
ax.set_xlabel('len')
plt.savefig('len.png')
with open('pr9/domain_organisation_list.txt', 'r') as names:
names_list = [i.strip() for i in names.readlines()]
table = pandas.read_csv('pr9/kakto.csv', sep='\t')
table['True'] = 'no'
table['1-spec'] = 0
table['sens'] = 0
table['F1'] = 0
table.loc[table['Sequence'].isin(names_list), 'True'] = 'yes'
for i in range(len(table)):
sl = table.loc[:i-1]
anti_sl = table.loc[i:]
yes_count = sl[sl['True'] == 'yes'].shape[0]
no_count = sl[sl['True'] == 'no'].shape[0]
anti_no = anti_sl[anti_sl['True'] == 'no'].shape[0]
table.loc[i, '1-spec'] = 1 - (anti_no+792-len(table))/(no_count+anti_no+792-len(table))
table.loc[i, 'sens'] = yes_count/len(names_list)
table.loc[i, 'F1'] = (2*yes_count)/(i+yes_count+no_count+1)
table.to_csv('pr9/kakto.csv', sep='\t', index=False)
fig = plt.figure()
ax = fig.add_subplot(1, 1, 1)
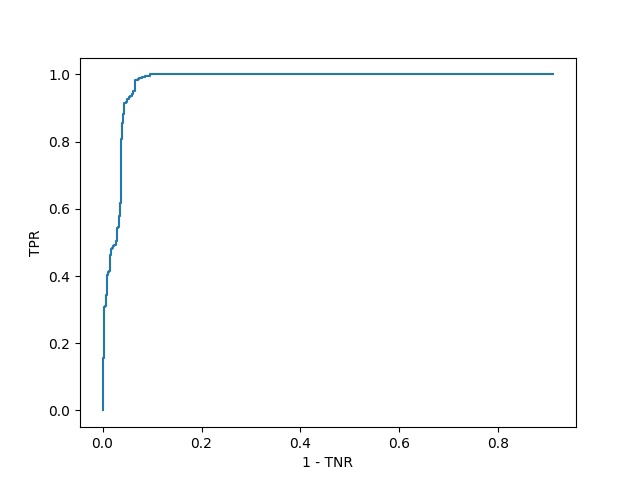
ax.plot( table['1-spec'], table['sens'], color='tab:blue')
ax.set_xlabel('1 - TNR')
ax.set_ylabel('TPR')
plt.savefig('ROC.png')
plt.close()
fig = plt.figure()
ax = fig.add_subplot(1, 1, 1)
numb = [i for i in range(len(table))]
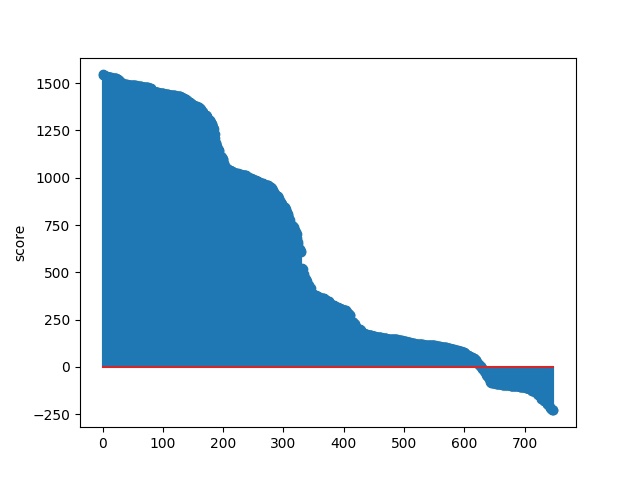
ax.stem(numb, table['score'])
ax.set_ylabel('score')
plt.savefig('score.png')
plt.close()
fig = plt.figure()
ax = fig.add_subplot(1, 1, 1)
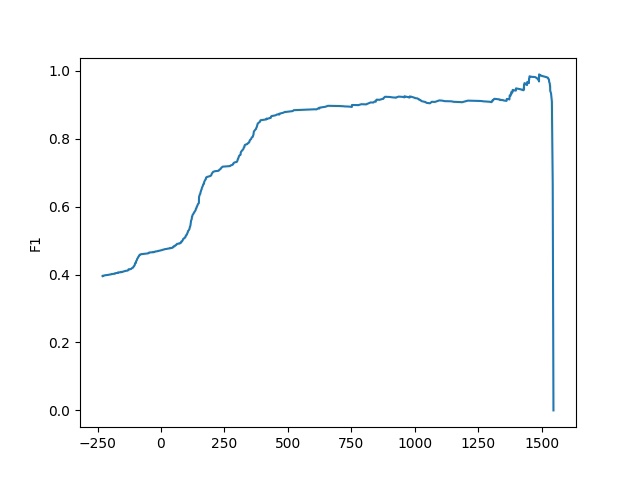
ax.plot(numb, table['F1'])
ax.set_ylabel('F1')
plt.savefig('F1.png')
plt.close()
Получены следующая таблица и графики
На Рис.1 видно 2 пика, первый из которых соответствует одно- и двухдоменной архитектурам. На Рис.2 и Рис.3 видны "волны", соответствующие белкам с разной архитектурой. График функции F1(Рис.4), также имеет "волны" в областях, соответствующейих скачку score между белками с разными архитектурами.