Практикум 12
Сравнение выравнивания одних и тех же последовательностей тремя разными программами
Проект Jalview с тремя выравниваниями
Полученные выравнивания в формате FASTA:
1) MUSCLE
2) MAFFT
3) T-coffee
Для получения сравнений выравниваний использовался код, написанный моей однокурсницей Гончаровой Еленой
совпадающие участки
| № | Muscle | Mafft |
|---|---|---|
| 1 | (1,32) | (1,32) |
| 2 | (42,45) | (42,45) |
| 3 | (54,57) | (54,57) |
| 4 | (63,93) | (64,94) |
| 5 | (237,252) | (376,391) |
| 6 | (286,288) | (444,446) |
| 7 | (290,315) | (448,473) |
несовпадающие участки
| № | Muscle | Mafft |
|---|---|---|
| 1 | (33,41) | (33,41) |
| 2 | (46,53) | (46,53) |
| 3 | (58,62) | (58,63) |
| 4 | (94,236) | (95,375) |
| 5 | (253,285) | 392,443) |
| 6 | (289,289) | (447,447) |
| 7 | (316,326) | (474,485) |
При сравнении выравниваний MUSCLE и MAFFT было обнаружено 7 блоков (119 колонок) одинаково выровненных колонок. Наличие относительно длинных блоков (максимальная длина — 32 а.к.о.) свидетельствует о высокой степени схожести этих двух алгоритмов друг с другом. Также совпадающие блоки в превой половине распалагаются в одинаковых местах выравниваний, что может свидетельствовать о том, что N-концевая часть белка эволюционно более консервативна, либо это связано со спецификой работы программ (так как в начале выравнивания гэпов еще мало, поэтому они не успели "сдвинуть" номера).
совпадающие участки
| № | Muscle | T-coffee |
|---|---|---|
| 1 | (1,10) | (1,10) |
| 2 | (13,29) | (13,29) |
| 3 | (41,45) | (41,45) |
| 4 | (53,59) | (53,59) |
| 5 | (63,93) | (63,93) |
| 6 | (237,258) | (472,493) |
| 7 | (264,266) | (499,501) |
| 8 | (290,319) | (529,558) |
несовпадающие участки
| № | Muscle | T-coffee |
|---|---|---|
| 1 | (11,12) | (11,12) |
| 2 | (30,40) | (30,40) |
| 3 | (46,52) | (46,52) |
| 4 | (60,62) | (60,62) |
| 5 | (94,236) | (94,471) |
| 6 | (259,263) | (494,498) |
| 7 | (267,289) | (502,528) |
| 8 | (320,326) | (559,565) |
В паре Muscle и T-coffee обнаружено 8 блоков совпадения (129 колонок). Максимальная длина блока составляет 31 а.к.о. В первой половине также наблюдаются совпадающие блоки на одинаковых позициях.
Совпадение Muscle с Mafft составило 36,5%, а Muscle с T-coffee - 39,6%. Результаты сравнения программ Muscle и Mafft, Muscle и T-coffee (такие как количество совпавших колонок и блоков, максимальная длина блока и др.) очень похожи друг на друга, поэтому можно предположить, что эти программы в данном случае взаимозаменяемы.
Построение выравнивания по совмещению структур и сравнение его с выравниванием программой MSA
Проект Jalview с двумя выравниваниями
Полученные выравнивания в формате FASTA:
совпадающие участки
| № | Muscle | PDBeFold |
|---|---|---|
| 1 | (21,99) | (25,103) |
| 2 | (118,156) | (121,159) |
| 3 | (159,199) | (163,203) |
несовпадающие участки
| № | Muscle | PDBeFold |
|---|---|---|
| 1 | (1,20) | (1,24) |
| 2 | (100-117) | (104,120) |
| 3 | (157,158) | (160,162) |
| 4 | (200,207) | (204,211) |
Одиночных совпадений вне блоков: 1 (1,1)
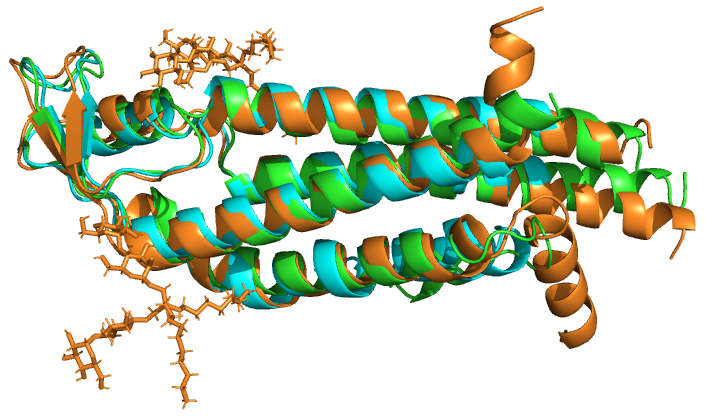
Оранжевый - 6l3u:A
Зеленый - 2zw3:A
Голубой - 6mhq:A
Были проанализированы выравнивания 3х последовательсностей А-цепей белков из домена Connexin (PF00029): 2ZW3, 6L3U, 6MHQ . Выравнивания имеют высокий процент сходства. Процент совпадающих колонок в выравнивании MUSCLE: 77,3%. Процент совпадающих колонок в выравнивании PDBeFold: 75,8% . Присутсвуют 3 больших совпадающих блока, длина которых составляет 79, 41 и 39 а.о. Из этого можно сделать вывод, что MUSCLE корректно выровнял последовательности и полученное выравнивание близко к истинному структурному выравниванию (структурные единицы более эволюционно устойчивы). Консервативные участки последовательности соответствуют структурно-консервативным участкам.
Программа MSA - MUSCLE
MUSCLE (от англ. Multiple Sequence Comparison by Log-Expectation) — компьютерная программа для множественного выравнивания белковых и нуклеотидных последовательностей. Разработана Робертом Эдгаром и впервые представлена в 2004 году в журнале Nucleic Acids Research [1].
Алгоритм включает быструю оценку попарных расстояний с помощью подсчёта k-меров, прогрессивное выравнивание с использованием целевой функции на основе логарифмического ожидания и итеративное уточнение с помощью зависимого от дерева перестроения профилей [1]. Оптимизированный алгоритм позволяет обрабатывать большие наборы данных (сотни последовательностей) за минуты. [2]
Преимущества перед аналогами
1) В 10–100 раз быстрее ClustalW и на порядки быстрее ProbCons [1, 2].
2) Для умеренно гомологичных последовательностей (идентичность >25%) точность MUSCLE достигает ~75%, что выше, чем у ClustalW (64,4%) [2].
Недостатки:
1) Менее точен при идентичности <15–20%.[2]
2) Плохо выравнивает участки, встречающиеся менее чем в 20% последовательностей.[2]
3) При количестве >80 последовательностей точность MUSCLE заметно снижается [2].
4) Не использует 3D-структуру белков в отличие от методов типа 3D-Coffee [1].
Источники
1) Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput //Nucleic acids research. – 2004. – Т. 32. – №. 5. – С. 1792-1797.
2) hompson J. D. et al. A comprehensive benchmark study of multiple sequence alignment methods: current challenges and future perspectives //PloS one. – 2011. – Т. 6. – №. 3. – С. e18093.