ПРАКТИКУМ 13
Сравнение выравниваний одних и тех же последовательностей разными программами
Для этого практикума, как и для предыдущих были выбраны последовательности с доменом PF00006, отвечающие ATP synthase alpha/beta family, nucleotide-binding domain. Множественное выравнивание было осуществленно, с помощью программ Mafft, Muscle и ClustalO. Muscle и ClustalO сравниванились относительно Mafft. Сравнение проводилось с помощью программы MACHO, написанной моими однокурсниками (Гагарочкин В., Масленников В., Нагорный Д.). В выбранном мною домене есть сильно выбивающиеся последовательности, поэтому, чтобы не учитывать блоки с большим количеством гэпов, несущие мало полезной информации, я ограничил количество гэпов в совпадающих блоках 10 штуками.
1. Сравнение выравниваний программы Mafft и ClustalO: 1.(536,539)=(522,525); 2.(543,545)=(529-531); 3.(548,549)=(534,535);
4.(565,574)=(551,560); 5.(580,596)=(566,582); 6.(607,610)=(593,596); 7.(629,630)=(613,614); 8.(634,651)=(618,635); 9.(737,746)=(710,719)
Схожесть выравниваний 2-х программ, если учитывать в совпадающих блоках все гэпы:
Длина первой последовательности: 746
Длина второй последовательности: 719
Доля совпавших колонок для выравнивания 1: 63.81 %
Доля совпавших колонок для выравнивания 2: 66.20 %
Одинаково выровненные колонки, которые я не включил в блоки, из-за большого количества гэпов: 1.(80,466)=(48,434); 2.(483,493)=(451,461); 3.(501,506)=(469,474); 4.(556,557)=(542,543)
2. Сравнение выравниваний программ Mafft и Muscle: 1.(1,31)=(1,31); 2.(535,545)=(532,542); 3.(580,595)=(586,601); 4.(608,616)=(614,622);
5.(629,630)=(633,634); 6.(633,651)=(637,655); 7.(724,727)=(722,725); 8.(737,746)=(735,744)
Схожесть выравниваний 2-х программ, если учитывать в совпадающих блоках все гэпы:
Длина первой последовательности: 746
Длина второй последовательности: 744
Доля совпавших колонок для выравнивания 1: 74.93 %
Доля совпавших колонок для выравнивания 2: 75.13 %
Одинаково выровненные колонки, которые я не включил в блоки, из-за большого количества гэпов: 1.(80,533)=(77,530); 2.(656,657)=(665,666); 3.(706,706)=(704,704)
Сравнение выравнивания по совмещению структур и программамой ClustalO
Для данного задания были выбраны белки, относящиеся к семейству PF00006, а именно альфа субъединицы АТФсинтазы с AC A0A009HWU4, A0A010RS60, A0A010SEU1.
Сперва было проведено совмещение структур и по нему сделано множественное выравнивание, потом для тех же белков была использована программа MSA ClustalO. Выравнивания получились почти идентичными, за исключением первых 65 аминокислотных остатков. В выравнивание по структуре гэпов оказалось больше, поэтому в выравнивание ClustalO было чуть больше консервативных участков. Различие в длине аминокислотных последовательностей хорошо прослеживается при сравнение пространственных структур белков. Видно, что у белка с AC A0A010RS60 есть дополнительный хвост, отходящий от бета-бочки.

Рисунок 1. Отличие в выравниваниях (верхнее - выравнивание структур; нижнее - выравнивание ClustalO)
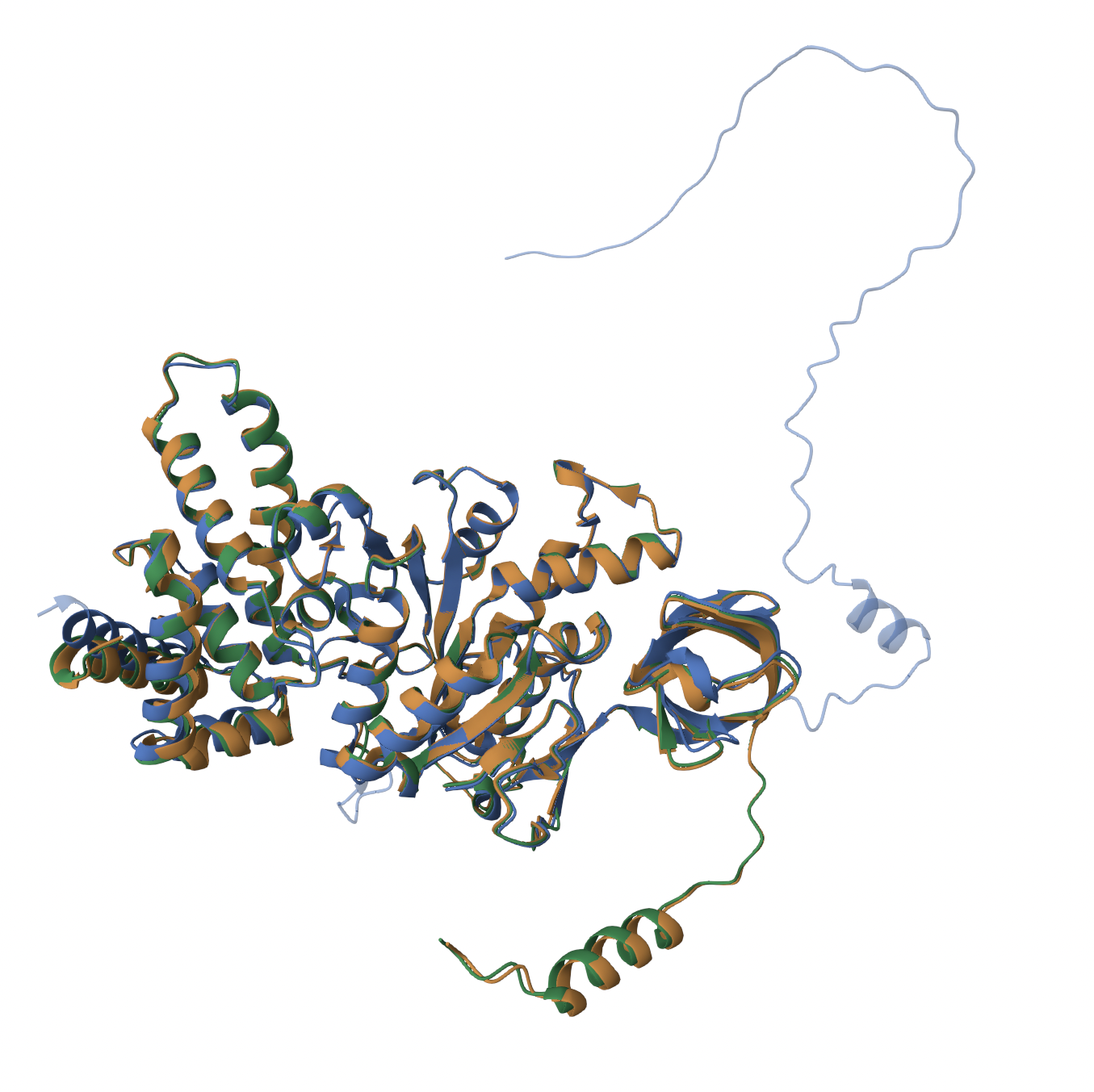
Рисунок 2. Сравнение пространственных структур
Краткое описание программы ClustalO
ClustalO (Clustal Omega) - программа для множественного выравнивания аминокислотных и нуклеотидных последовательностей.
Представляет из себя современную версию программы Clustal (Cluster alignment). В случае небольшого количества выравниваемых последовательностей
программа показывает схожие результаты по точности и скорости с программами Mafft и Muscle, однако на очень больших группах последовательностей, эта программа имеет преимущества, за счет использования моделей HMM.
Общий план работы:
- K-tuple distance matrix: Все последовательности разбиваются на небольшие фрагменты, эти фрагменты подсчитываются, после чего строится матрица растояний, показывающая насколько две последовательности отличаются
- Initial guide tree: На основе матрицы строиться направляющее дерево методом UPGMA, показывающее в каком порядке и с кем надо объединить последовательность
- Initial Alignment: Пошаговое выравнивание, когда сперва выравниваются самые схожие последовательности и постепенно, основываясь на guide tree к ним добавляются новые последовательности
- HMM: В случае если последовательностей очень много, то дополнительно используются скрытые марковские модели (HHM), они позволяют представить промежуточные выравнивания как HMM-профиль и уже дальше сравнивать их, что многократно повышает скорость выравнивания
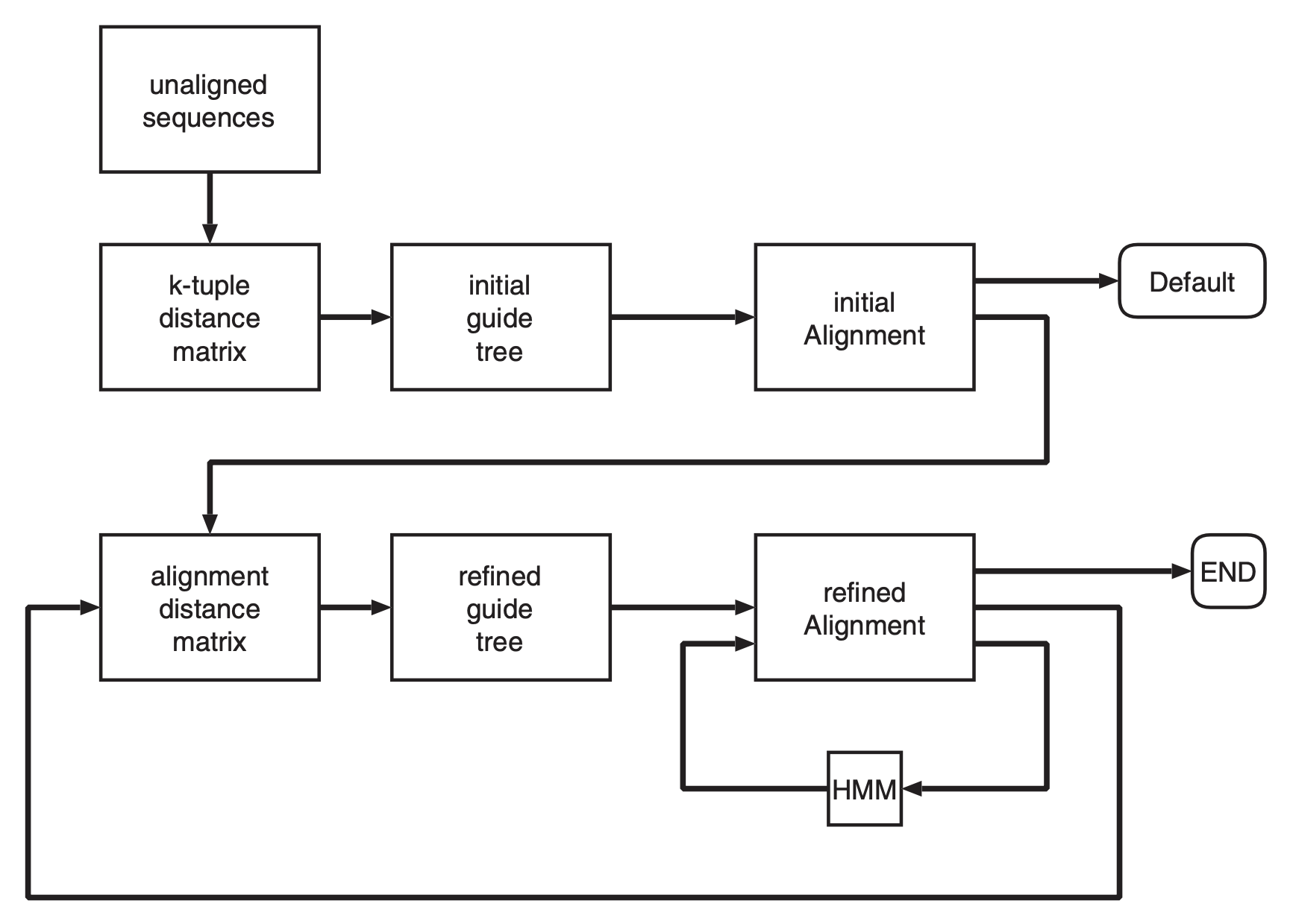
Рисунок 3. Принцип работы программы ClustalO
Литература:
[1] Sievers, F., Higgins, D. G. 2014. Clustal Omega. Curr. Protoc. Bioinform. 48:3.13.1-3.13.16.
doi: 10.1002/0471250953.bi0313s48
[2] David J. Russell (ed.), Multiple Sequence Alignment Methods, Methods in Molecular Biology, vol. 1079, DOI 10.1007/978-1-62703-646-7_6, © Springer Science+Business Media, LLC 2014