Практикум 12. Алгоритмы и программы множественного выравнивания
Сравнение выравнивания одних и тех же последовательностей разными вариантами программ MSA
Сравнение множественных выравниваний нескольких последовательностей домена PF00374 (из выравнивания seed), рассмотренного в практикуме 11, было проведено для программ Muscle, Mafft и Clustal в Jalview "ручным" способом (без использования специализированных программ). Выравнивания Muscle и Mafft совпадают в следующих блоках:
- (1,99)=(1,99)
- (103,121)=(103,121)
- (127,235)=(127,235)
- (239,259)=(239,259)
- (271,288)=(271,288)
- (295,304)=(295,304)
- (311,400)=(311,400)
- (405,543)=(405,543)
Для Muscle и Clustal список одинаковых блоков другой:
- (1,99)=(1,99)
- (103,121)=(103,121)
- (127,233)=(127,233)
- (242,265)=(242,265)
- (271,288)=(271,288)
- (294,306)=(294,306)
- (317,338)=(316,337)
- (344,397)=(343,396)
- (405,512)=(404,511)
- (517,543)=(516,542)
Таким образом, выравнивания Muscle и Mafft различаются на 38 колонок, а Muscle и Clustal - на 52 или 51 (в зависимости от того, по какому выравниванию считать), то есть выравнивания Muscle и Mafft более сходны друг с другом, чем Muscle и Clustal, хотя в целом они достаточно сильно похожи и отличаются только в низкоконсервативных областях. Можно объяснить это тем, что из 3 алгоритмов Clustal был разработан раньше всех и может уступать более современным программам по точности.
Результаты в Jalview доступны по ссылке, также можно скачать множественные выравнивания в fasta-формате: Muscle, Mafft и Clustal.
Построение выравнивания по совмещению структур и сравнение его с выравниванием MSA
Выравнивание по совмещению структур было построено с помощью PDBeFold для 3 белков с доменом PF00374 - гидрогеназы-1 E.coli (код в PDB - 3uqy), NiFe-гидрогеназы Desulufovibrio vulgaris (1wul) и NiFe-гидрогеназы Desulfovibrio desulfuricans (1e3d); его визуализация представлена на рис.1. Можно заметить, что укладка 3 белков очень похожа, и основные элементы вторичной структуры почти полностью накладываются, а различия есть только в положении неструктурированных участков.
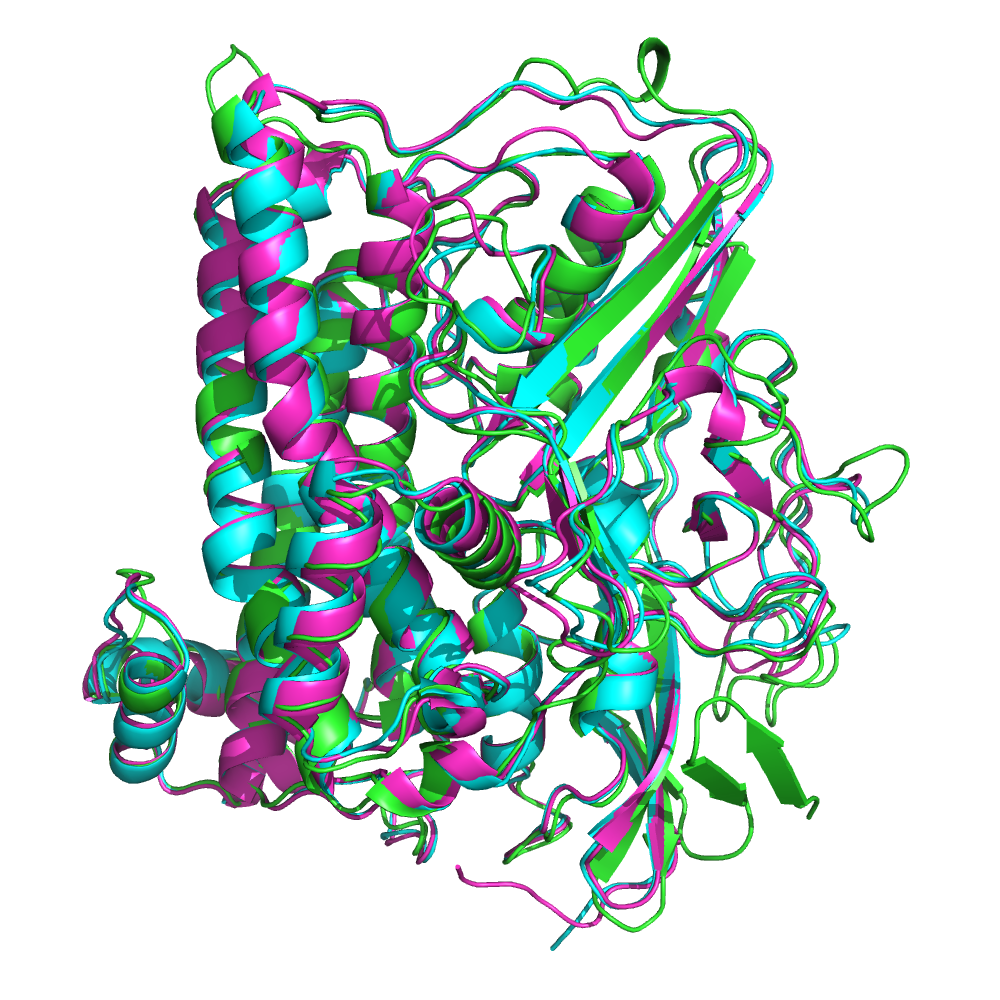
Также последовательности были выровнены программой Muscle в Jalview. При сравнении этих выравниваний (способом, аналогичным тому, что применялся в предыдущем задании) выделяются следующие совпадающие блоки (сначала колонки в выравнивании по совмещению структур, затем - в Muscle):
- (17,36)=(17,36)
- (40,153)=(40,153)
- (157,232)=(157,232)
- (235,241)=(235,241)
- (260,296)=(259,295)
- (303,311)=(302,310)
- (323,341)=(320,338)
- (349,359)=(346,356)
- (366,380)=(363,367)
- (398,483)=(395,480)
- (501,587)=(498,584)
- Кроме того, совпадает также 486 (483) колонка.
Из сравнения выравниваний можно сделать вывод, что они выявляют одни и те же консервативные участки, которые соответствуют элементам вторичной структуры, в то время как участки, не имеющие определённой укладки, являются неконсервативными и выравниваются плохо. В частности, значительно различаются выравнивания N-концевого участка.
Ссылки: проект Jalview, выравнивание по структуре и выравнивание Muscle в fasta-формате.
Описание программы MSA
Clustal Omega - современный алгоритм множественного выравнивания последовательностей, который был представлен в 2011 году. Как и другие алгоритмы Clustal, он использует метод прогрессивного выравнивания с использованием направляющего дерева. Clustal Omega позволяет составлять выравнивания из очень большого количеста последовательностей за счёт особенно эффективного алгоритма построения направляющего дерева mBed, в котором каждая последовательность заменяется вектором с n координат, каждая из которых представляет собой расстояние до другой последовательности, после чего эти векторы кластеризуются стандартными методами. Ещё одним нововведением в ClustalO стало использование скрытых марковских моделей (HMM), с помощью которых строится итоговое выравнивание. Для некоторых целей, таких как, например, выравнивание последовательностей белковых доменов, можно использовать HMM, полученную на основе уже сущетствующего выравнивания, в качестве "внешнего профиля". Всё это делает ClustalO достаточно точным и быстрым алгоритмом, имеющим преимущество на больших наборах последовательностей.
Список литературы
Sievers, Fabian, et al. "Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega." Molecular systems biology 7 (2011): 539.