
1. Описание файла с референсом
Мне досталась 8-я хромосома человека. После открытия сразу бросается в глаза огромное количество неизвестных нуклеотидов. Путём несложных операций, было подсчитано, что их 375986 (примерно 0,25%). Также я подсчитал сколько оснований каждого типа содержится в файле: А - 146535711, Т - 146532483, G - 146486382, C - 146490999. Тогда соотношение A:T:G:C будет примерно 1:1:1:1 (с точностью до десятитысячных).
2. Индексация референса
При помощи команды bwa index -a bwtsw chr9.fna была проведена индексация. Здесь параметр "-a" отвечает за создание BWT индекса (преобразование Барроуза-Уиллера для сжатия данных).
Изначально у него есть два варианта: "is" и "bwtsw". Мы использовали 2-й (SW - алгоритм выравнивание Смита-Ватермана), так как этот метод адаптирован для работы с целым человеческим геномом.
3. Описание образца
- Ссылка на образец в БД NCBI
- Прибор: Illumina Genome Analyzer IIx
- Организм: Homo sapiens
- Стратегия секвенирования: whole-exome
- Тип чтений: парноконцевые
- Ожидаемое кол-во чтений: 41,482,410
4. Проверка качества исходных чтений
При помощи fastqc были проанализированы полученные чтения.
- fastqc SRR10720414_1.fasta.gz - ссылка
- fastqc SRR10720414_2.fasta.gz - ссылка
Во всех картинках сначала идёт последовательность reverse, а затем forward
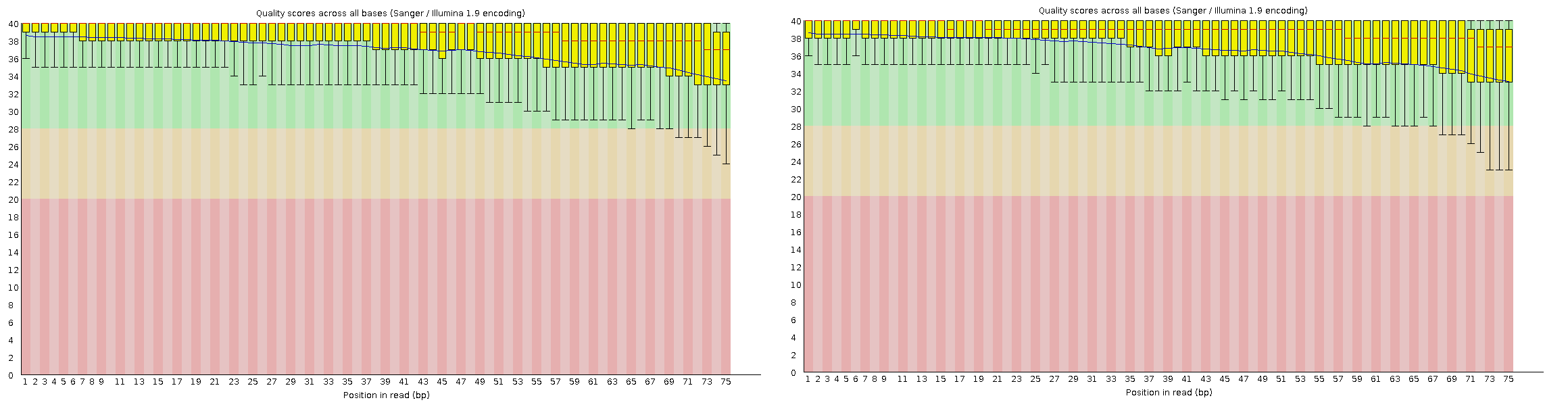
Так как изображение достаточно большое, то вот ссылка, чтобы его скачать.
Итого получилось 41482410 чтений (как и ожидалось), причём как у прямых, так и у обратных. Качество получилось отличное (среднее примерно 32). В самом конце качество довольно сильно ухудшается, что логично.
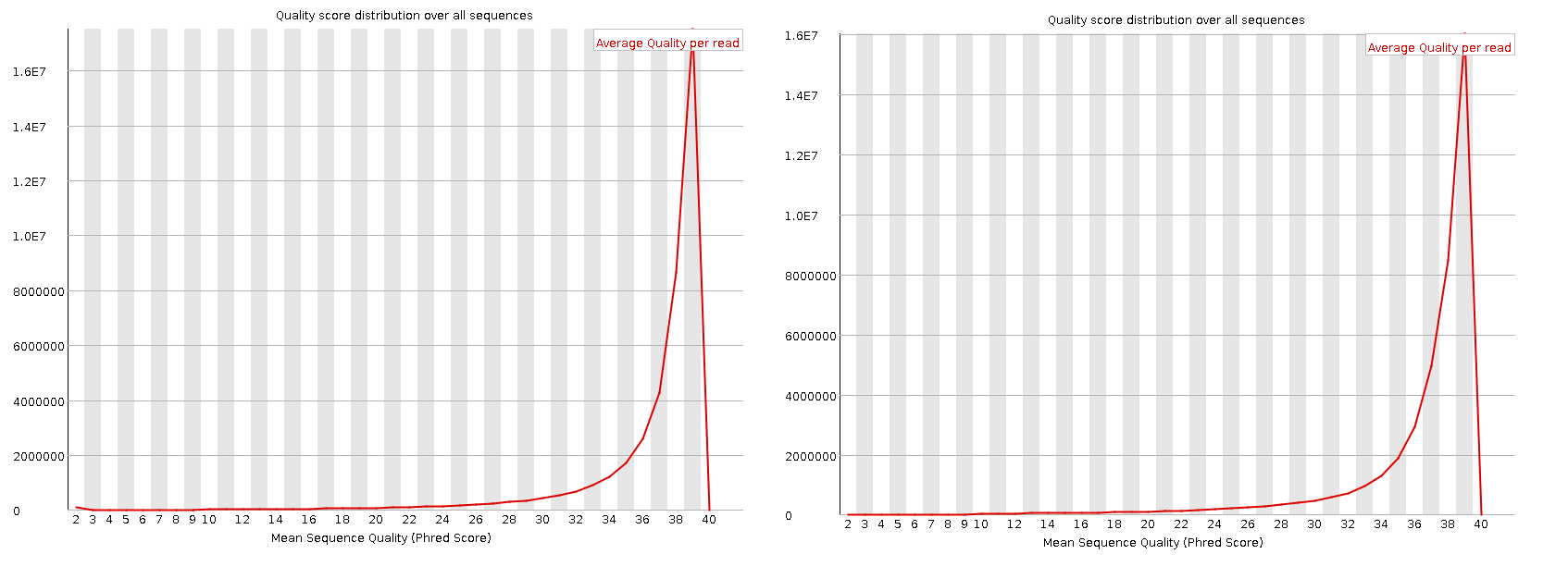
Так как изображение достаточно большое, то вот ссылка, чтобы его скачать.
Видно, что получилось хорошее распределение, так как все последовательности формируют один очень узкий пик с неизменно высоким баллом. В плохом случае получился бы ещё пик, который означал бы, что есть последовательности, которые имеют в целом более низкое качество. Тогда надо бы было отфильтровать набор последовательностей, но у нас всё хорошо.
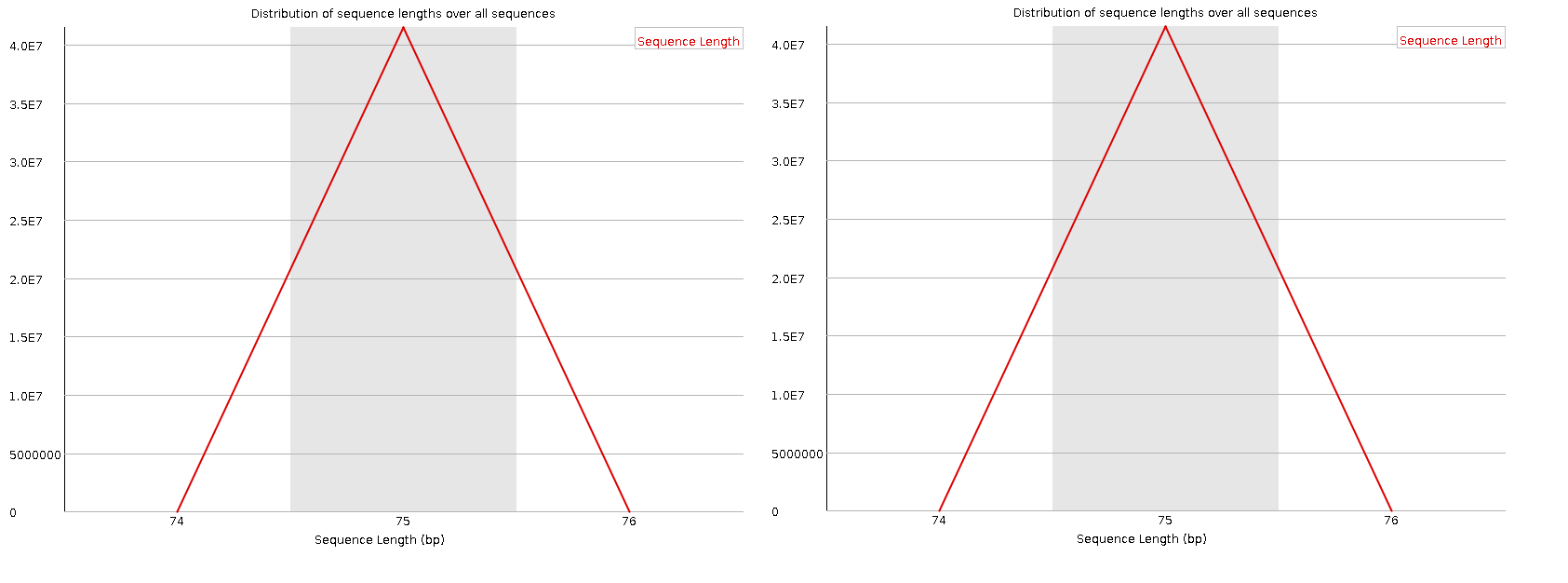
Так как изображение достаточно большое, то вот ссылка, чтобы его скачать.
Данный график показывает распределение длин фрагментов в файле (например, чтобы посмотреть, что у нас не получилось всё одинаковой длины). Наибольшее количество последовательностей с длиной 75, что объясняет такое гигантское число прочтений, но зато увеличивается точность.
5. Фильтрация чтений
При помощи алгоритма Trimmomatic был проведён тримминг чтений. Итоговая команда выглядела так:
java -jar /usr/share/java/trimmomatic.jar PE -threads 10 -phred33 -trimlog trimlog_dna ../dna_data/SRR10720414_1.fastq.gz ../dna_data/SRR10720414_2.fastq.gz dna_f_p.fastq.gz dna_f_u.fastq.gz dna_r_p.fastq.gz dna_r_u.fastq.gz TRAILING:20 MINLEN:50
А теперь, объясним параметры. Прибор кодирует качество в формате phred-score=33. Trailing=20 позволяет удалить нуклеотиды с конца чтений с качеством ниже 20. Minlen=50 удаляет все чтения с длиной меньше 50. Threads=10 "выдаёт на пользование" 10 ядер, чтобы не ждать слишком долго.
6. Проверка качества триммированных чтений
При помощи всё того же fastqc:
- fastqc dna_f_p.fastq.gz - ссылка
- fastqc dna_r_p.fastq.gz - ссылка
- fastqc dna_f_u.fastq.gz - ссылка
- fastqc dna_r_u.fastq.gz - ссылка
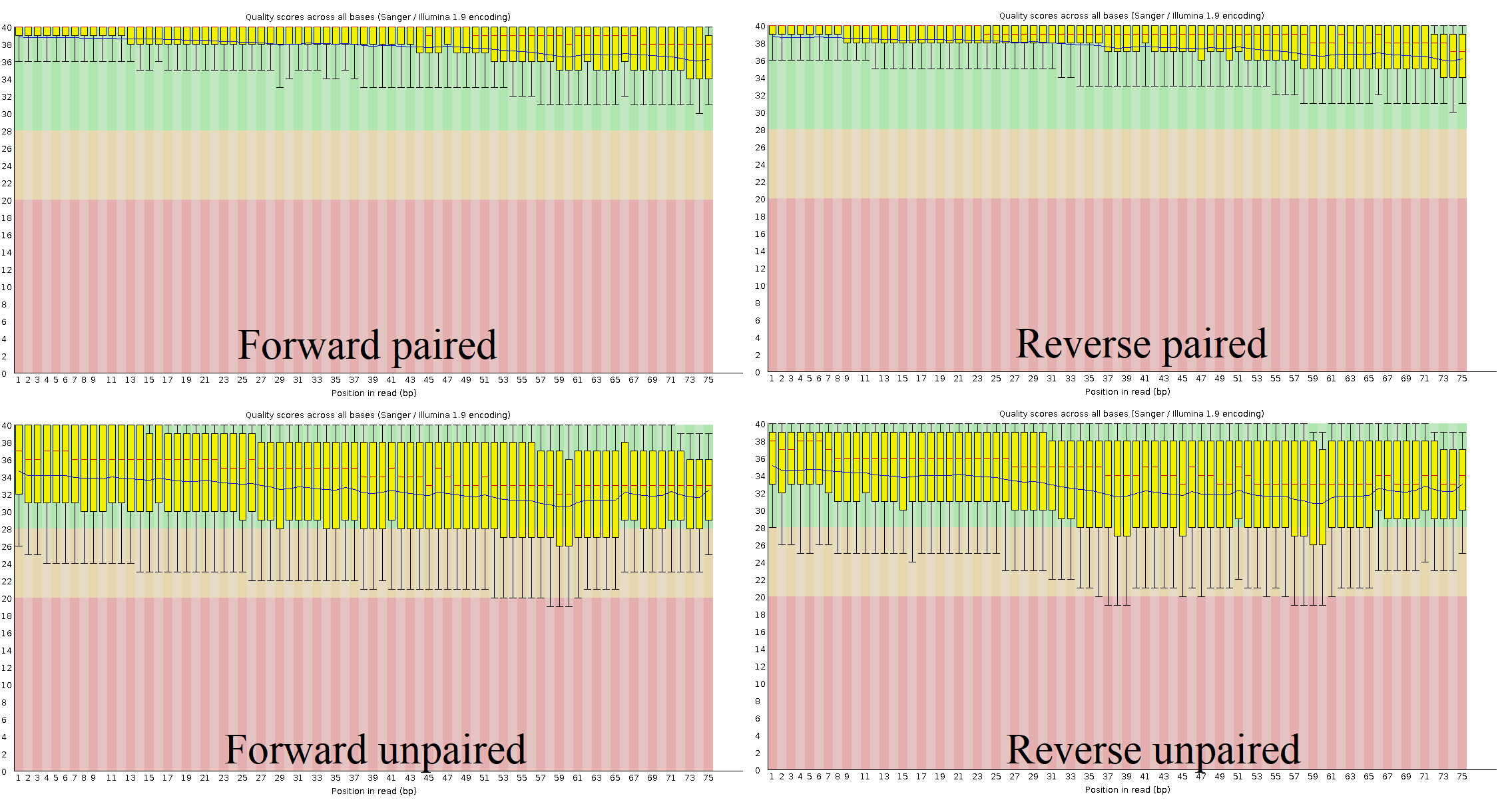
В прямом и обратном парных прочтениях осталось 39888569 (96.16%) чтений. Из предложенной картинки видно, что качество неспаренных чтений ощутимо ухудшилось. Возможно, это объясняется тем, что парные чтения дополняют друг друга и уменьшают вероятность получения ошибки. Парные прочтения же теперь лежат полностью в зелёной части графика (примерное среднее 35).
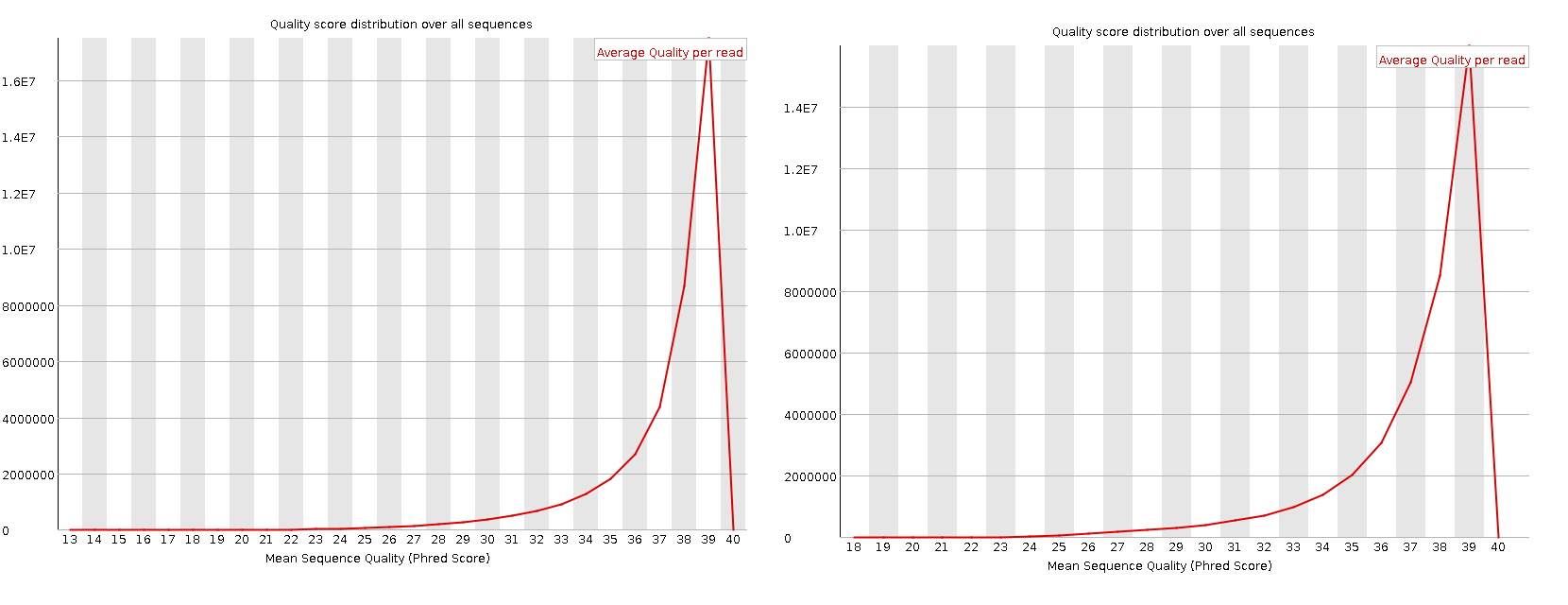
Далее предлагается работать только с парными прочтениями. На предоставленном графике показано распределение счёта (score) последовательностей. Так как мы убрали слишком короткие прочтения, то общий счёт увеличился. То есть большее количество последовательностей имело больший счёт, что прижало график слева к нижней оси и увеличило ширину пика (ну и к тому же последовательностей стало поменьше и масштаб изменился).
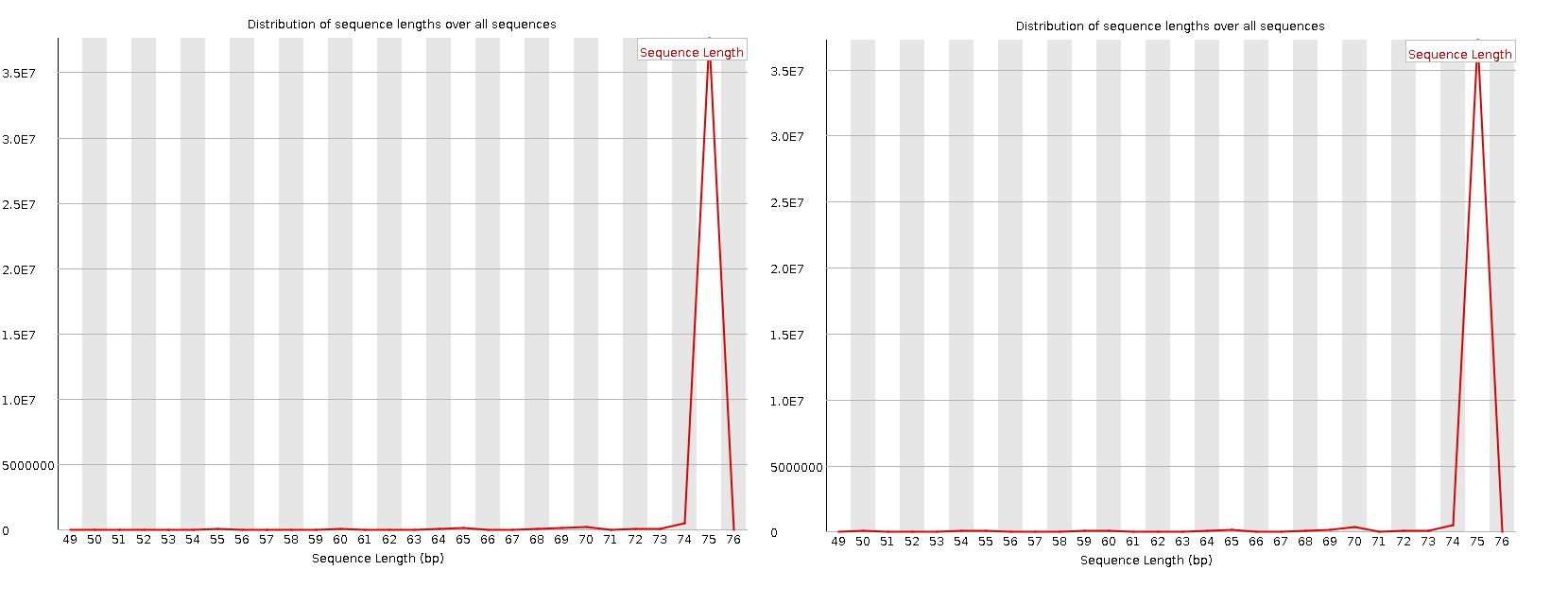
Выше представлено распределение длин последовательностей. Так как мы удалили все последовательности, длиной меньше 50, то и отсчёт теперь сдвинулся. Пик всё также находится на 75.