 8 (926) 907 94 08
8 (926) 907 94 08
Всё на свете является чудом!
Эволюционные домены
Подготовка к работе
Для выполнения работы по дальнейшей реконструкции эволюции доменной архитектуры белков был выбран белок THIS_BACSU, выданный ещё во втором семестре I курса. Структура этого белка содержит один единственный домен ThiS, который характерен для целого семейства белков.
Описание домена
Белки из семейства ThiS (thiaminS) участвуют в передаче серы, имеют два консервативных глицина на конце COOH. Тиокарбоксилат формируется на последнем глицине в процессе активации. Сера передается от ThiI к ThiS в реакции, катализируемой ISCS. MoaD участвует в передаче серы в синтезе молебнопротеина и показывает ограниченное сходство последовательностей к ThiS.
Pfam AC - PF02597
Pfam ID - ThiS
Ниже представлена небольшая таблица, в которой сравниваются данные о домене с прошлого года и то, что мы имеем на данный момент:
| Год | Количество архитектур в которые входит домен | Количество доступных последовательностей | Количество белков, для которых определена пространственная структура PDB | Всего найденных структур PDB |
| 2012 | 30 | 4665 | 13 | 28 |
| 2013 | 34 | 6781 | 17 | 44 |
Как мы видим, база данных немного пополнилась за прошедший год. Так что динамика развития банка есть, и это не может не радовать.
Работа с последовательностями
Используя программу JalView, загрузили выравнивание из Pfam (File > Fetch Sequences по идентификатору PF02597) и раскрасили его по консервативности (ClustalX и By conservation с порогом консервативности 10%).
Затем из 17 белков, для которых определена структура PDB выбрали один - CYSO_MYCTU (3DWM), и, выделив все последовательности, сделали по ним поиск этого белка, после чего добавили к найденной последовательности 3D структура (PDB ID - 3DWM).
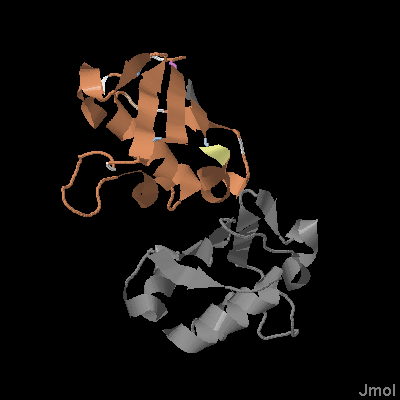 Слева приведено изображение структуры.
Слева приведено изображение структуры.И, наконец, сохранили проведённую работу как проект JalView,
а также как выравнивание в fasta-формате.
Если проект JalView не откроется напрямую, то через File>Load Project откроется точно.
2. Выбор архитектур
Получение информации об архитектуре всех последовательностей, содержащих выбранный домен
Используя скрипт swisspfam-to-xls.py и файл swisspfam-2011.gz, содержащий информацию об архитектуре всех последовательностей UniProt, преобразовали информацию о доменах в последовательности из файла swisspfam в таблицу для Excel (по умолчанию result.xls):
python swisspfam-to-xls.py -i /srv/databases/pfam/swisspfam-2011.gz -z -p PF02597
В итоге, как и планировали, получили таблицу с информацией об архитектуре последовательностей, содержащих домен ThiS (лист "Main page", колонки зелёного цвета), которую потом сохранили как сводную таблицу (лист "Summary table").
Получение информации о таксономической принадлежности и длине домена
По идентификаторам отобранных последовательностей в UniProt были получены AC (ID Mapping), по которым был получен файл со списком AC в формате UniProt (Retrieve) [из исходных 4907 идентификаторов после сопоставления осталось 4572 - потери небольшие, поэтому выяснять причины отсутствия остальных идентификаторов не стали], который затем был использован для работы скрипта uniprot-to-taxonomy.py :
python uniprot-to-taxonomy.py -i flat_text.txt -o flat_text_out.txt
В результате получили файл с таксономией, информацию из которого добавили на отдельный лист "Taxonomy" книги Excel, а также с помощью функции ВПР перенесли полученную таксономию в основную таблицу (лист "Main page", колонки голубого цвета).
Также был добавлен столбец, содержащий информацию о длине домена ThiS из каждой последовательности (лист "Main page", колонка красного цвета).
Выбор архитектур
Для дальнейшего изучения эволюции доменных архитектур, включающих домен ThiS, были выбраны две двухдоменные архитектуры (ThiS, MoaE) и (ThiS, ThiG). Ниже представлена таблица с информацией об этих архитектурах:
| № | Домены | Число представителей | Описание других доменов |
| 1 |
This, MoaE (PF02391) |
138 | MoaE: участвует в биосинтезе молибдоптерина, который является универсальным компонентом кофакторов птеринового молибдена, а также содержит дитиол группы, служащие для привязки MoaE. Добавление дитиол серы к молибдоптерину требует активность преобразовывающего фактора. Преобразовывающий фактора содержит в себе MoaE и MoaD. |
| 2 |
This, ThiG (PF05690) |
33 | ThiG: вместе с ThiF и ThiH принимает участие в синтезе 4-метил-5-(б-гидроксиэтил)-тиазола (THZ), который является промежуточным продуктом на пути производства тиазола. |
3. Выбор таксона
Выбор таксона
Чтобы изучить выбранные архитектуры были выбраны 2 крупных таксона Bacteria и Archaea, так как в более мелких таксонах нужного количества последовательностей нет.
Для архитектуры [This, MoaE] выбрали подтаксоны: Thermoprotei (из таксона Archaea); Actinobacteria, Bacilli и Deltaproteobacteria (из таксона Bacteria).
Для архитектуры [This, ThiG] выбрали подтаксоны: Clostridia, Alphaproteobacteria и Gammaproteobacteria (из таксона Bacteria).
4. Выбор представителей архитектур
Из каждой архитектуры отобрали последовательности (40 для первой архитектуры [32 после выбрасывания некоторых] и 25 для второй архитектуры [15 после выбрасывания некоторых]). Результат отбора представлен на листе "Selected Seqs" в книге Excel (последовательности также отмечены цветом на листе "Summary table"). Затем они были использованы, чтобы оставить в выравнивании нужные последовательности из двух групп. Для этого воспользовались скриптом filter_alignment.py:
python filter_alignment.py -i domains_this.fasta -m ids_selected_domains.txt -o filter_alignment_out.fasta
Полученное выравнивание было загружено в JalView и отредактировано (удалены пустые колонки). Затем в нём были созданы и названы 2 группы согласно архитектуре, в каждой из них была выполнена раскраска последовательностей ClustalX, Conservation (были выбраны пороги на консервативность 15% и 20% для первой и второй групп соответственно). После повторного редактирования выравнивания (удалены несодержательные N- и C-концевые участки) в файле выравнивания осталось 47 последовательностей.
5. Построение филогенетического дерева
Чтобы построить филогенетическое дерево сначала отредактируем имена последовательностей таким образом, чтобы в названии каждой из них была отражена доменная архитектура и сравниваемые таксоны. Для сравнения были выбраны две двухдоменные архитектуры, поэтому закодируем их просто 1 и 2. Соответствующие выбранные таксоны были обозначены следующим образом:
• для доменной архитектуры 1 - ThiS & MoaE:
- T - Thermoprotei
- Ac - Actinobacteria
- B - Bacilli
- D - Deltaproteobacteria
• для доменной архитектуры 2 - ThiS & ThiG:
- C - Clostridia
- Al - Alphaproteobacteria
- G - Gammaproteobacteria
Для того, чтобы переименовать последовательности в файле с выравниванием, были использованы файл с информацией о таксономии белков, в которых присутствует домен ThiS, и файл с информацией об архитектуре последовательностей, и применён скрипт rename_seqs.py.
Построение дерева на основе полученного выравнивания было выполнено методами UPGMA (предполагает молекулярные часы, строит укоренённое дерево) и Neighbor-Joining (не использует гипотезу о молекулярных часах и строит неукоренённое дерево), чтобы затем иметь возможность сравнить построенные деревья. Полученные с помощью программы MEGA изображения построенных деревьев приведены ниже (Neighbor-Joining слева и UPGMA справа). Использовали бутстреп анализ, чтобы точнее определить правильность ветвей по ей поддержке (чем ближе число к 100, тем выше поддержка).
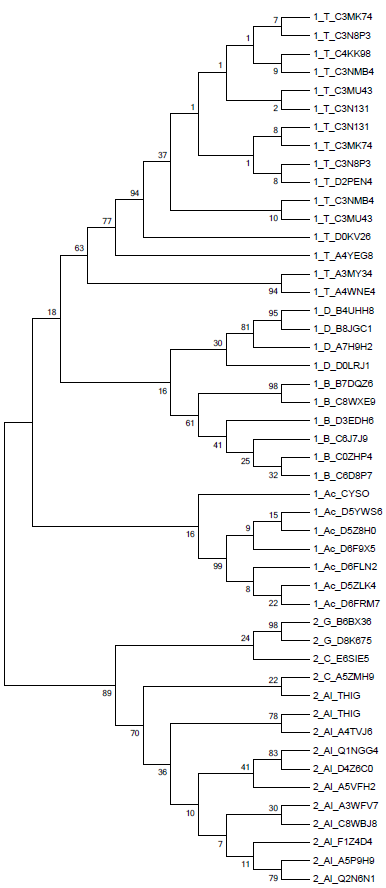
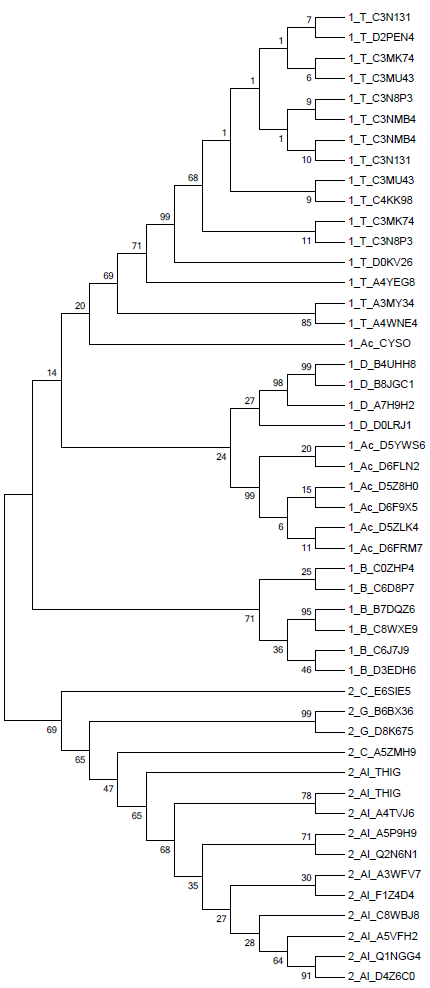
Лучше с задачей справился метод Neighbor-Joining, так как UPGMA допустил ошибку, поместив под общую ветвь подтаксона Thermoprotei белок 1_Ac_CYSO, тогда как Neighbor-Joining поместил этот белок согласно таксономии вместе с остальными представителями подтаксона Actinobacteria.
Ниже приведено изображение, где выделена поддержка нужных ветвей, а также показаны доменные архитектуры:
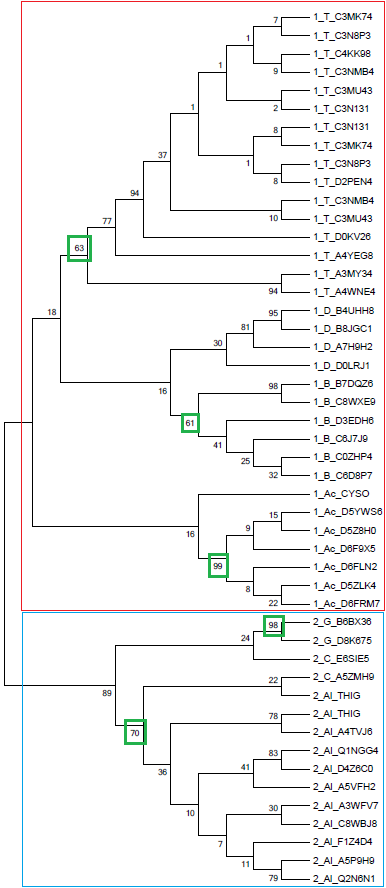
Нужные ветви - это такие ветви на дереве, которые совпадают с ветвями по таксономии.
На дереве выше у этих ветвей (T-Thermoprotei, B-Bacilli, Ac-Actinobacteria, G-Gammaproteobacteria, Al-Alphaproteobacteria) зелёным цветом обведена их поддержка.
Ветвь D-Deltaproteobacteria тоже отделена, но имеет высокую поддержку только в ветвях низшего порядка, а у самой ветви поддержка всего 30, что означает не совсем правильное расположение ветви на дереве (заметим, что методом UPGMA тоже не удалось построить дерево с высокой поддержкой этой ветви - поддержка всего 27).
Далее воспользовались программой визуализации деревьев ITOL:
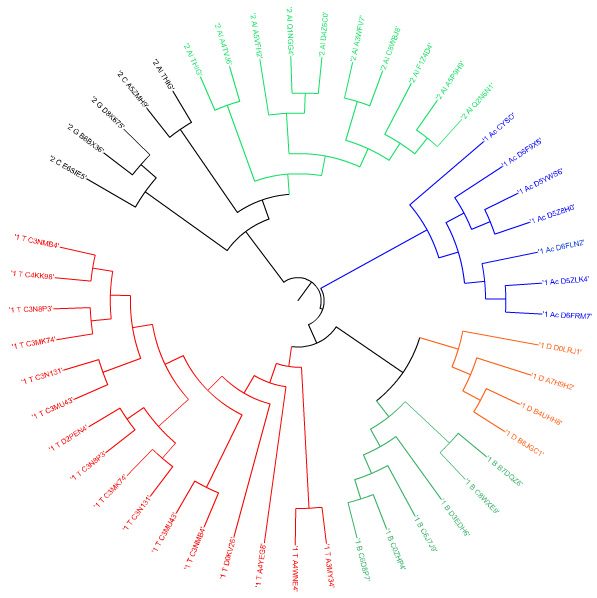
Здесь уже отчётливо выделяются подтаксоны Thermoprotei (Кренархеоты), Actinobacteria (Актинобактерии), Bacilli (Бациллы), Deltaproteobacteria (Дельта-протеобактерии) и Alphaproteobacteria (Альфа-протеобактерии), поэтому можно выносить суждения об их эволюции.
Изначально два рассматриваемых таксона - Архей и Бактерий - объединяли в один и называли прокариотами, но со временем установили, что археи имеют свою независимую эволюционную историю и характеризуются многими биохимическими особенностями, отличающими их от других форм жизни.
Всё это понятно, но на деле (на дереве) мы видим вот какую картину: Бациллы и Дельта-протеобактерии действительно имеют общего предка и находятся под общей ветвью, но Альфа-протеобактерии, имеющие большее родство с Дельта-бактериями, чем Бациллы, хоть и находятся под общей ветвью, но слишком высшего порядка. Актинобактерии более менее грамотно заняли место в дереве, с учётом эволюции.
Но это всё мелочи, ведь эти подтаксоны относятся к общему таксону Bacteria, как и должны. Основной загвоздкой в отображении эволюции на дереве встал подтаксон Кренархеоты, который вместо эволюционно приписанного таксона Архей, находится под ветвью Бактерий. И выходит, что на дереве есть только один таксон - Бактерии.
Единственным объяснением произошедшего является то, что в геномах архей и бактерий могли происходить трансдукции, трансформации и другие виды горизонтального переноса (при участии бактериофагов и др. способом). При этом получается, что в геноме организма оказываются чужеродные гены, последовательности которых вполне могут оказаться на дереве, представленном выше.
Что касается двух выбранных архитектур, на продублированном рисунке дерева, мы видим, что они обе изначально отделились друг от друга, и уже потом произошло независимое разделение последовательностей в каждой архитектуре на подтаксоны. С правильным разделением архитектур справились оба метода реконструкции деревьев (NJ и UPGMA).
| Главная | Об авторе | Учебные семестры | Проекты автора | Друзья | Ссылки партнеров | Extra | Контакты |
Mneff © 2011-2013